
Bioenergetika dědičných metabolických poruch
Významnou skupinou dědičných mitochondriálních poruch jsou defekty ATP syntázy, klíčového enzymu buněčného energetického metabolizmu. Detailně proto zkoumáme sestavování tohoto proteinového komplexu složeného z mnoha podjednotek a proteiny, které se jej účastní.
Studium pacientů s mitochondriálními onemocněními
V mitochondriích probíhá celá řada důležitých metabolických drah včetně oxidativní fosforylace (OXPHOS), beta oxidace mastných kyselin a Krebsova cyklu. Není tedy překvapující, že mitochondriální dysfunkce může mít za následek řadu lidských patologií.
Výzkum lidských mitochondriálních patologií, který využívá vzorků z takto postižených pacientů, významně přispěl k objevu nových mitochondriálních proteinů a jejich funkcí. Uvážíme-li, že podle současných odhadů existuje přibližně 300 mitochondriálních proteinů, které dosud vůbec nebyly identifikovány, může výzkum v této oblasti přispět k objasnění mnoha nemocí.
Dnes charakterizace nového pacienta typicky začíná exomovým sekvenováním, což patří mezi specializace našeho spolupracujícího pracoviště – Kliniky dětského a dorostového lékařství (KDDL) 1. Lékařské fakulty UK v Praze. Naší doménou pak je následná biochemická a funkční charakterizace nově identifikovaných proteinů.
Biogeneze ATP syntázy
ATP syntáza je enzym, kterému se věnujeme již mnoho let. Snažíme se poznat geny, jejichž mutace vedou k lidským patologiím, stejně tak jako charakterizovat dráhy vedoucí k sestavování tohoto enzymu složeného z mnoha podjednotek.
V roce 2008 jsme se spolupodíleli na objasnění genetické příčiny mitochondriálního onemocnění velké skupiny pacientů. Podařilo se totiž najít homozygotní mutaci v jaderném genu Tmem70. Přesná funkce jím kódovaného proteinu TMEM70 není dosud známá, ale zřejmě se účastní sestavování proteinového komplexu ATP syntázy. U pacientů totiž dochází k významnému snížení množství kompletně sestavených komplexů ATP syntázy (Čížková et al., 2008).
V současnosti se snažíme protein TMEM70 blíže charakterizovat. Zjistili jsme, že z 29-kDa prekurzoru je po importu do mitochondrií odštěpena N-koncová mitochondriální signální sekvence. Maturovaný protein (21 kDa) je lokalizovaný ve vnitřní mitochondriální membráně (Hejzlarová et al., 2011).
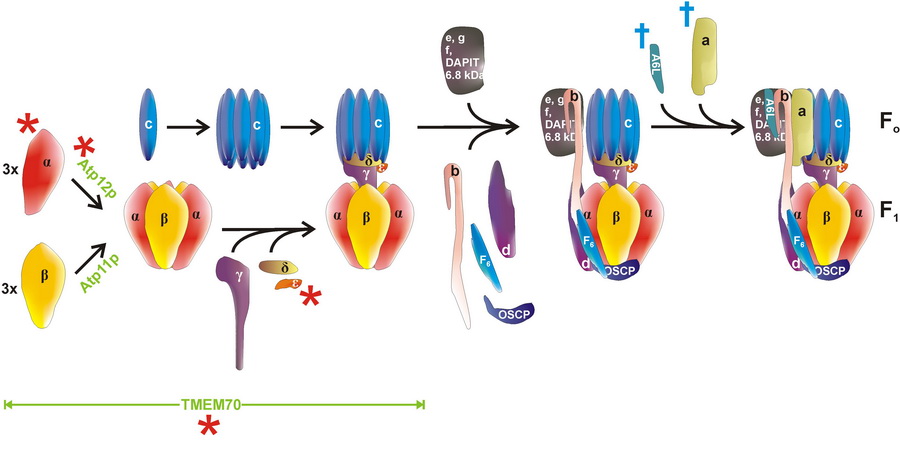
Schéma postupného sestavování komplexu ATP syntázy v savčích mitochondriích.
Současné projekty zahrnují:
- Úloha proteinu TMEM70 v biogenezi ATP syntázy (myší knock-out genu Tmem70 – ve spolupráci s dr. Sedláčkem, ÚMG AV ČR; potkaní knock-out genu Tmem70 – ve spolupráci s dr. Pravencem, FGÚ AV ČR).
- Úloha podjednotek centrálního stonku ATP syntázy v biogenezi tohoto enzymu (buněčné linie s knock-downovanými geny kódujícími podjednotky γ, δ a ε).
- Hledání nových proteinů asociovaných s ATP syntázou nebo regulujících její biogenezi.
Superkomplexy mitochondriálních enzymů
Studujeme asociaci enzymů OXPHOS do vyšších strukturních celků, tzv. superkomplexů. Jedná se o rychle se rozvíjející oblast, v rámci níž se soustředíme především na výzkum strukturních interakcí tzv. flavinových dehydrogenáz s jinými komponenty OXPHOS, případně dalších mitochondriálních metabolických drah (Krebsův cyklus).