
Our new publication: High resistance to mitochondrial mutations
A developing organism requires high amounts of energy in the form of ATP. In higher eukaryotes, and thus in humans, more than 90 % of ATP is produced in mitochondria, a key organelle of the cellular catabolism. It is therefore not surprising that mitochondrial defects belong to the most frequent causes of metabolic diseases in children.
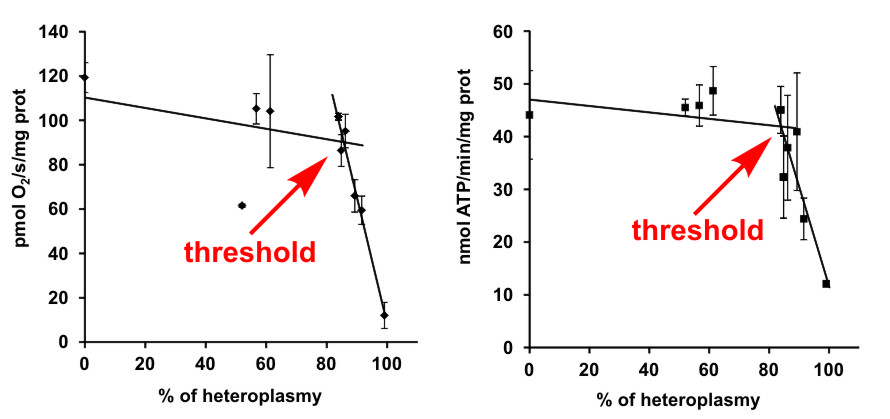
ATP synthase and other enzymes of the oxidative phosphorylation system share one unique trait, namely the dual genetic origin of their subunits, which are encoded by DNA from the nucleus and also by DNA in mitochondria (mtDNA). Both nuclear and mitochondrial DNA mutations may be manifested as hereditary diseases. Clinical symptoms of mtDNA patients usually vary from mild muscle dysfunctions to very severe heart and brain defects. Many different symptoms can be found also in patients with the same mutation, even in the siblings where the nuclear background has a minimal influence. The main factor affecting the disease severity is the so-called heteroplasmy that expresses the percentage of mutated mtDNA in respect to the healthy, wild-type mtDNA. This year, we published a study performed on the model of a rare mutation in the MT-ATP6 gene, leading to a defect of two oxidative phosphorylation enzymes: cytochrome c oxidase and ATP synthase. Although these are key enzymes for ATP production in the mammalian cell, we showed that a high threshold level of mutation load above 90 % of heteroplasmy is necessary to be reached for the biochemical manifestation of the mutation and thus for the development of severe symptoms. The aim of the study was to shed light on the relationship between the mutation load (level of heteroplasmy) and its clinical manifestation. In this specific case, the severity of symptoms is directly linked with the residual amount of the respective enzyme subunits, and the threshold originates from the gene/protein level.
Hejzlarová K, Kaplanová V, Nůsková H, et al. Alteration of structure and function of ATP synthase and cytochrome c oxidase by lack of Fo-a and Cox3 subunits caused by mitochondrial DNA 9205delTA mutation. Biochem. J. 2015; 466:601–611.