
Naše nová publikace: Vysoká odolnost buněk vůči mitochondriálním mutacím
Vyvíjející se organismus potřebuje velké množství energie ve formě ATP. U vyšších eukaryot, a tedy i u člověka, je více než 90 % potřebného ATP produkováno v mitochondriích, které obecně představují klíčovou organelu buněčného katabolismu. Není tedy překvapivé, že mitochondriální poruchy patří mezi časté příčiny metabolických onemocnění u dětské populace.
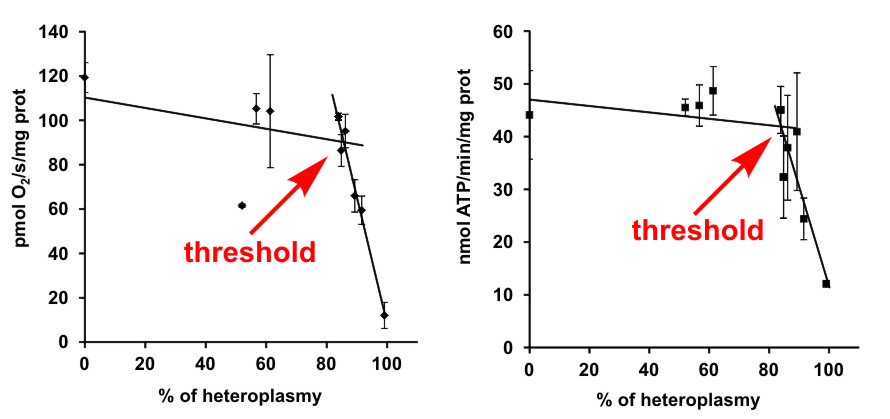
ATP syntáza sdílí s dalšími enzymy oxidační fosforylace jednu unikátní vlastnost, a sice že její podjednotky jsou kódovány jak DNA v jádře, tak i DNA obsaženou přímo v mitochondriích (mtDNA). Mutace v jaderné i mitochondriální DNA se pak mohou manifestovat jako dědičná onemocnění. Klinické obrazy u mtDNA pacientů bývají různé, od lehčích svalových dysfunkcí až po velmi závažná onemocnění mozku a srdce. I u pacientů se stejnou mutací je možné nalézt řadu rozdílných symptomů, a to i u sourozenců, kde je vliv jaderného pozadí minimální. Hlavním faktorem ovlivňujícím závažnost těchto onemocnění je takzvaná heteroplazmie, která vyjadřuje procentuální zastoupení mutované mtDNA relativně ke zdravé, nemutované. V letošním roce jsme opublikovali studii provedenou na modelu vzácné mutace v mitochondriálním genu MT-ATP6, která vede k defektu hned dvou enzymů oxidační fosforylace: cytochrom c oxidázy a ATP syntázy. Ukázali jsme, že i přesto, že se jedná o dva klíčové enzymy tvorby ATP v savčí buňce, pro biochemický projev mutace a rozvoj závažných příznaků onemocnění je nezbytné, aby obsah mutované mitochondriální DNA překročil velmi vysokou prahovou hodnotu heteroplazmie okolo 90 %. Snahou publikace bylo přispět právě k objasnění vztahu mezi zastoupením mutace (hladinou heteroplazmie) a jejími klinickými příznaky. V tomto konkrétním případě se ukázalo, že závažnost projevů přímo odpovídá množství přítomných podjednotek jednotlivých enzymů a ke vzniku prahového efektu dochází na úrovni překladu gen/protein.
Hejzlarová K, Kaplanová V, Nůsková H, et al. Alteration of structure and function of ATP synthase and cytochrome c oxidase by lack of Fo-a and Cox3 subunits caused by mitochondrial DNA 9205delTA mutation. Biochem. J. 2015;466:601–611.