Insights into the operational model of agonism of receptor dimers

Jakubík, J.; Randáková, A. Insights into the Operational Model of Agonism of Receptor Dimers. Expert Opin. Drug Discov. 2022, 17, 1181–1191, doi:10.1080/17460441.2023.2147502. Invited Review.
- The exact ranking of efficacies and potencies of agonists is indispensable in the discovery of new selective agonists.
- The operational model of agonism (OMA) is the current standard.
- Many receptors function as oligomers, requiring an extension of the classical OMA.
- Extension of OMA by slope factors gives simple equations of functional response that are easy to fit experimental data but results may be inaccurate.
- Extension of OMA by cooperativity factors gives accurate but complex equations of functional response.
- Extension of OMA by cooperativity factors should be preferred.
Introduction: Accurate ranking of efficacies and potencies of agonists is essential in the discovery of new selective agonists. For the purpose of system-independent ranking of agonists, the operational model of agonism (OMA) has become a standard. Many receptors function as oligomers which makes functional responses more complex, requiring an extension of the original OMA.
Areas covered: Explicit equations of the operational model of agonism of receptor dimers (OMARD) were derived. The OMARD can be applied to any receptor possessing two orthosteric sites. The behaviour of OMARD was analysed to demonstrate its complexity and relation to experimental data. Properties of OMARD and OMA equations were compared to demonstrate their pros and cons.
Expert opinion: Extension of OMA by slope factors gives simple equations of functional response that are easy to fit experimental data but results may be inaccurate. Also, such equations cannot accommodate bell-shaped curves. Explicit equations of OMARD give accurate results but are complex and tedious to fit experimental data. All operational models use inter-dependent parameters that are a hurdle in the fitting.
Functionally selective and biased agonists of muscarinic receptors.

Biased signaling at muscarinic receptors
Randáková, A.; Jakubík, J. Functionally Selective and Biased Agonists of Muscarinic Receptors. Pharmacol. Res. 2021, 169, 105641, doi:10.1016/j.phrs.2021.105641.
Invited review.
Disruption of cholinergic signalling via muscarinic receptors is associated with various pathologies, like Alzheimer’s disease or schizophrenia. Selective muscarinic agonists possess therapeutic potential in the treatment of diabetes, pain or Sjögren’s syndrome. The orthosteric binding site of all subtypes of the muscarinic receptor is structurally identical, making the development of affinity-based selective agonists virtually impossible. Some agonists, however, are functionally selective; they activate only a subset of receptors or signalling pathways. Others may stabilize specific conformations of the receptor leading to non-uniform modulation of individual signalling pathways (biased agonists). Functionally selective and biased agonists represent a promising approach for the selective activation of individual subtypes of muscarinic receptors. In this work, we review chemical structures, receptor binding and agonist-specific conformations of currently known functionally selective and biased muscarinic agonists in the context of their intricate intracellular signalling. Further, we take a perspective on the possible use of biased agonists for tissue and organ-specific activation of muscarinic receptors.
Nicotinic Acetylcholine Receptors Expressed by Striatal Interneurons Inhibit Striatal Activity and Control Striatal-Dependent Behaviors.

Behavioral tests performed in individual cohorts of mice. The order of tests in each cohort and the approximate age of tested animals are shown. We indicate each month of age with an arrow; each arrow can host 4 dots representing the 4 weeks of the month.
Abbondanza, A.; Ribeiro Bas, I.; Modrak, M.; Capek, M.; Minich, J.; Tyshkevich, A.; Naser, S.; Rangotis, R.; Houdek, P.; Sumova, A.; et al. Nicotinic Acetylcholine Receptors Expressed by Striatal Interneurons Inhibit Striatal Activity and Control Striatal-Dependent Behaviors. J. Neurosci. 2022, 42, 2786–2803, doi:10.1523/JNEUROSCI.1627-21.2022.
A large variety of nicotinic acetylcholine receptors (nAChRs) are expressed in the striatum, a brain region that is crucial in the control of behaviour. The complexity of receptors with different functions is hindering our understanding of mechanisms through which striatal acetylcholine modulates behaviour. We focused on the role of a small population of beta2-containing nAChRs. We identified neuronal types expressing these receptors and determined their impact on the control of explorative behaviour, anxiety-like behaviour, learning, and sensitivity to stimulants. Additional experiments showed that these alterations were associated with an overall increased activity of striatal neurons. Thus, the small population of nicotinic receptors represents an interesting target for modulation of response to stimulant drugs and other striatal-based behaviour.
Acetylcholine is an important modulator of striatal activity, and it is vital to controlling striatal-dependent behaviours, including motor and cognitive functions. Despite this significance, the mechanisms determining how acetylcholine impacts striatal signalling are still not fully understood. In particular, little is known about the role of nAChRs expressed by striatal interneurons. In the present study, we used FISH to determine which neuronal types express the most prevalent beta2 nicotinic subunit in the mouse striatum. Our data support a common view that nAChR expression is mostly restricted to striatal interneurons. Surprisingly though, cholinergic interneurons were identified as a population with the highest expression of beta2 nicotinic subunit. To investigate the functional significance of beta2-containing nAChRs in striatal interneurons, we deleted them by injecting the AAV-Cre vector into the striatum of beta2-flox/flox male mice. The deletion led to alterations in several behavioural domains, namely, to an increased anxiety-like behavior, a decrease in sociability ratio, a deficit in discrimination learning, and increased amphetamine-induced hyperlocomotion and c-Fos expression in mice with beta2 deletion. Further colocalization analysis showed that the increased c-Fos expression was present in both medium spiny neurons and presumed striatal interneurons. The present study concludes that, despite being relatively rare, beta2-containing nAChRs are primarily expressed in striatal neurons by cholinergic interneurons and play a significant role in behaviour.
Neurosteroids and steroid hormones are allosteric modulators of muscarinic receptors

Dolejší, E.; Szánti-Pintér, E.; Chetverikov, N.; Nelic, D.; Randáková, A.; Doležal, V.; Kudová, E.; Jakubík, J. Neurosteroids and Steroid Hormones Are Allosteric Modulators of Muscarinic Receptors. Neuropharmacology 2021, 199, 108798, doi:10.1016/j.neuropharm.2021.108798.
- Some neurosteroids and steroid hormones bind to muscarinic acetylcholine receptors with the affinity of 100 nM or greater
- Steroids acting at nanomolar concentrations represent the novel pharmacophore of allosteric modulators of muscarinic receptors
- Corticosterone and progesterone allosterically modulate muscarinic receptors at physiologically relevant concentrations
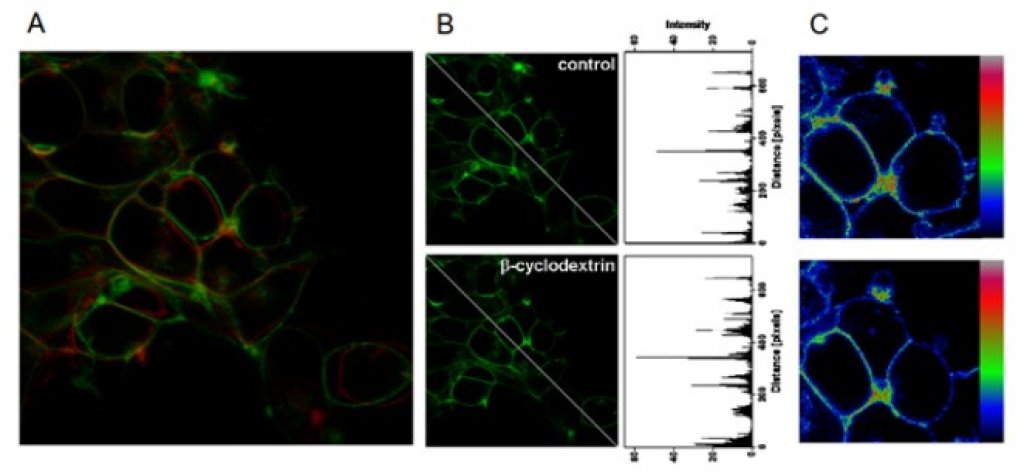
The membrane cholesterol was found to bind and modulate the function of several G-protein coupled receptors including muscarinic acetylcholine receptors. We investigated the binding of 20 steroidal compounds including neurosteroids and steroid hormones to muscarinic receptors. Corticosterone, progesterone and some neuro- steroids bound to muscarinic receptors with the affinity of 100 nM or greater. We show that corticosterone and progesterone allosterically modulate the functional response of muscarinic receptors to acetylcholine at physiologically relevant concentrations. It can play a role in stress control or in pregnancy, conditions where levels of these hormones dramatically oscillate. Allosteric modulation of muscarinic receptors via the cholesterol- binding site represents a new pharmacological approach to diseases associated with altered cholinergic signalling. Further, we established a structure-activity relationship for steroid-based allosteric modulators of muscarinic receptors.
The operational model of allosteric modulation of pharmacological agonism

The cubic ternary complex model of allosteric modulation of receptor activation. Inactive, R, and active, R*, states of the receptor, K<sub>ACT</sub> receptor activation constant, K<sub>A,/sub> and K<sub>B</sub> affinities.
Proper determination of agonist efficacy is indispensable in the evaluation of agonist selectivity and bias to activation of specific signalling pathways. The operational model of pharmacological agonism is a useful means for achieving this goal.
Monod et al. (1963) originally introduced the concept of allosterism. Since then the concept of allosterism extended to many various fields of research spanning from DNA expression via metabolism to ion channels and G-protein coupled receptors. Allosteric ligands bind to a site that is distinct from the orthosteric site on a receptor. An orthosteric and allosteric ligand can bind to the receptor concurrently and form a ternary complex where they reciprocally modulate the binding affinity of each other. Moreover, the binding of an allosteric modulator may also affect the efficacy of an orthosteric agonist in eliciting a functional response.
Allosteric modulators are an intensively studied group of receptor ligands because of their selectivity and preservation of physiological space-time pattern of the signals they modulate. We analysed the operational model of allosterically-modulated agonism (OMAM) including modulation by allosteric agonists. Several parameters of OMAM are inter-dependent. We derived equations describing mutual relationships among parameters of the functional response and OMAM. We present a workflow for the robust fitting of OMAM to experimental data using derived equations.
Novel M2-Selective, Gi-Biased Agonists of Muscarinic Acetylcholine Receptors

General scheme of coupling GPCRs with individual subtypes of G‑proteins or arrestins after activation by a traditional full agonist (left) and an agonist preferring Gi‑protein activation (right). AC, adenylyl cyclase; PLC, phospholipase C.
We have developed new muscarinic receptor agonists as a pharmacophore for the development of new non-addictive analgesics such as opiates or weakening of immunity as steroid analgesics. These muscarinic analgesics would not cause side effects such as incontinence, excessive salivation and sweating, and others.
More than 30 % of currently marketed medications act via G-protein coupled receptors (GPCRs). This type of membrane receptors is involved in the control of a wide range of physiological processes, from the processing of sensory stimuli, through the regulation of behaviour and mood, hormonal and immune responses, to the control of autonomic functions and cell proliferation. Thus, GPCRs represent one of the most important pharmacotherapeutic targets. In contrast to traditional agonists activating multiple signalling pathways, agonists biased towards a given signalling pathway represent a new generation of drugs with increased specificity and fewer adverse effects. Enormous research has been done on agonists biased towards either G-protein or arrestin mediated pathway.
In our laboratory, we focus on the study of muscarinic acetylcholine receptors, which belong to the typical representatives of GPCR. Here, as a proof of concept, we demonstrate unprecedented signalling bias solely at the level of G‑protein-mediated signalling. We present agonists of muscarinic acetylcholine receptors, a member of GPCR family, that exclusively inhibit cAMP synthesis through activation of Gi‑protein pathway and thus are functionally selective for M2 and M4 receptor subtypes. Muscarinic receptors M2, M4 represent one of the pharmacological targets in the treatment of pain. Newly discovered agonists may lead to the development of new non-addictive analgesics such as opiates, or immunity-weakening as steroid analgesics. Such muscarinic analgesics would not cause side effects mediated by activation of the Gq protein signalling pathway, such as incontinence, excessive salivation and sweating, and more.
Agonist-specific conformations of the M2 muscarinic acetylcholine receptor assessed by molecular dynamics

Structurally diverse agonists for a given receptor induce receptor conformations specific to each structural family. These agonist-specific conformations can lead to non-uniform modulation of signalling pathways. This preferential orientation of signalling of a given receptor towards a subset of its signal transducers is termed signalling bias. This property may be employed to develop drugs that selectively produce desired effects while avoiding side effects associated with activation of unwanted signalling pathways. We modelled conformations of the M2 receptor specific to individual agonists, including the newly developed Gi-biased agonists.
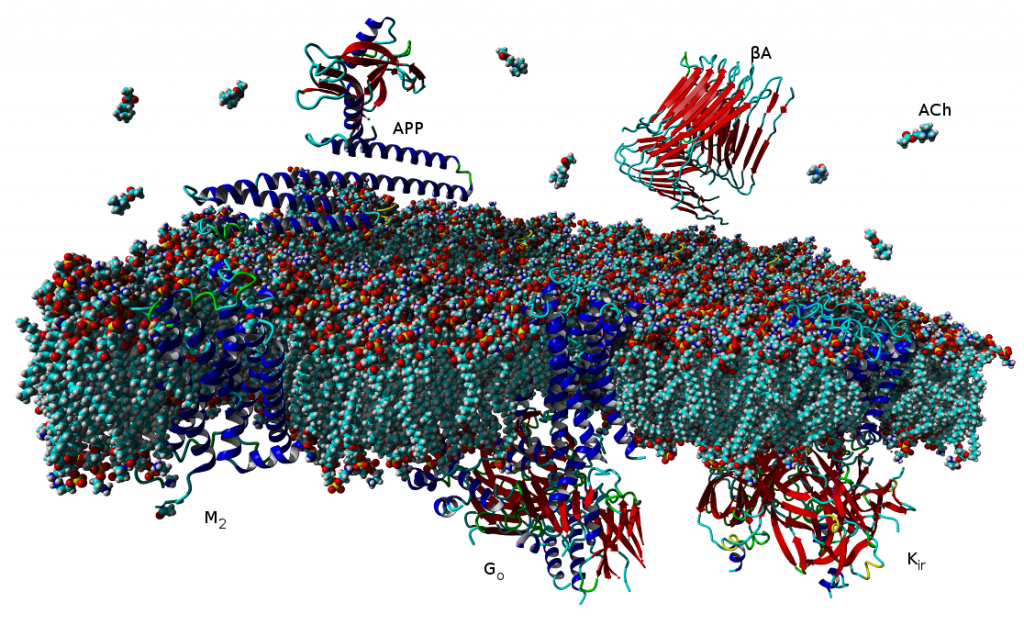
Binding of muscarinic ligands, both antagonists and agonists, and their effects on the conformation of the M2 acetylcholine receptor were modelled in silico and compared to experimental data. After docking of antagonists to the M2 receptor in an inactive conformation (3UON, 5ZK3, 5ZKB or 5ZKB) and agonists in an active conformation (4MQS) 100 ns of conventional molecular dynamics (MD) followed by 500 ns of accelerated MD was run. Conventional MD revealed ligand-specific interactions with the receptor. Antagonists stabilized the receptor in an inactive conformation during accelerated MD. The receptor in complex with various agonists attained different conformations specific to individual agonists. The magnitude of TM6 movement correlated with agonist efficacy at non-preferential Gs pathway. The shape of the intracellular opening where the receptor interacts with a G-protein was different for the classical agonist carbachol, super-agonist iperoxo and Gi/o-biased partial agonists JR-6 and JR-7, being compatible with experimentally observed agonist bias at the G-protein level. Moreover, wash-resistant binding of the unique agonist xanomeline associated with interaction with membrane lipids was formed during accelerated MD. Thus, accelerated MD is suitable for modelling of ligand-specific receptor binding and receptor conformations that is essential for the design of experiments aimed at the identification of the secondary binding sites and understanding molecular mechanisms underlying receptor activation.
Applications and limitations of fitting of the operational model to determine relative efficacies of agonists

Graphical Abstract
Proper determination of agonist efficacy is essential in the assessment of agonist selectivity and signalling bias. Agonist efficacy is a relative term that is dependent on the system in which it is measured, especially being dependent on receptor expression level. In this work, we analyse limits and pitfalls of fitting OM to experimental data.
Proper determination of agonist efficacy is essential in the assessment of agonist selectivity and signalling bias. Agonist efficacy is a relative term that is dependent on the system in which it is measured, especially being dependent on receptor expression level. The operational model (OM) of functional receptor agonism is a useful means for the determination of agonist functional efficacy using the maximal response to agonist and ratio of agonist functional potency to its equilibrium dissociation constant (KA) at the active state of the receptor. However, the functional efficacy parameter τ is inter-dependent on two other parameters of OM; agonist’s KA and the highest response that could be evoked in the system by any stimulus (EMAX). Thus, fitting of OM to functional response data is a tricky process. In this work, we analyse pitfalls of fitting OM to experimental data and propose a rigorous fitting procedure where KA and EMAX are derived from the half-efficient concentration of agonist and apparent maximal responses obtained from a series of functional response curves. Subsequently, OM with fixed KA and EMAX is fitted to functional response data to obtain τ. The procedure was verified at M2 and M4 muscarinic receptors fused with the G15 G-protein α-subunit. The procedure, however, is applicable to any receptor-effector system.
Role of membrane cholesterol in differential sensitivity of muscarinic receptor subtypes to persistently bound xanomeline

Our new publication in journal Neuropharmacology in which we demonstrate that membrane cholesterol plays an important and subtype-specific role in activation of muscarinic acetylcholine receptors. To our knowledge, this is the first demonstration of pharmacological selectivity due to differences in receptor-membrane interactions at any GPCR. The possibility to achieve pharmacological selectivity based on receptor-membrane interactions changes our view on the molecular basis of pharmacological selectivity and opens new ways for the development of a novel pharmaceutics.
Xanomeline (3-(Hexyloxy)-4-(1-methyl-1,2,5,6-tetrahydropyridin-3-yl)-1,2,5-thiadiazole) is a muscarinic agonist that is considered to be functionally selective for the M1/M4 receptor subtypes. Part of xanomeline binding is resistant to washing. Wash-resistant xanomeline activates muscarinic receptors persistently, except for the M5 subtype. Mutation of leucine 6.46 to isoleucine at M1 or M4 receptors abolished persistent activation by wash-resistant xanomeline. Reciprocal mutation of isoleucine 6.46 to leucine at the M5 receptor made it sensitive to activation by wash-resistant xanomeline. Lowering of membrane cholesterol made M1 and M4 mutants and M5 wild type receptors sensitive to activation by wash-resistant xanomeline. Molecular docking revealed a cholesterol binding site in the groove between transmembrane helices 6 and 7. Molecular dynamics showed that interaction of cholesterol with this binding site attenuates receptor activation. We hypothesize that differences in cholesterol binding to this site between muscarinic receptor subtypes may constitute the basis for xanomeline apparent functional selectivity and may have notable therapeutic implications. Differences in receptor-membrane interactions, rather than in agonist-receptor interactions, represent a novel possibility to achieve pharmacological selectivity. Our findings may be applicable to other G protein coupled receptors.

Cholesterol binding to the intracellular half of TM6 of wt (left) and L376I mutant (right) M1 receptor based on crystal structure 5CXV (Thal et al., 2016) is shown. Orientation, extracellular side up, TM6 front. Colours: magenta, surface of TM5, TM6 and TM7, yellow, surface of R365, L376 and I376; cyan, carbon; white, hydrogen; red, oxygen.
Novel long-acting antagonists of muscarinic ACh receptors

Graphical Abstract
In this study, we have developed new potent and long-acting antagonists of muscarinic acetylcholine receptors with a potential therapeutic use.
BACKGROUND AND PURPOSE:
The aim of this study was to develop potent and long-acting antagonists of muscarinic ACh receptors. The 4-hexyloxy and 4-butyloxy derivatives of 1-[2-(4-oxidobenzoyloxy)ethyl]-1,2,3,6-tetrahydropyridin-1-ium were synthesized and tested for biological activity. Antagonists with long-residence time at receptors are therapeutic targets for the treatment of several neurological and psychiatric human diseases. Their long-acting effects allow for reduced daily doses and adverse effects.
EXPERIMENTAL APPROACH:
The binding and antagonism of functional responses to the agonist carbachol mediated by 4-hexyloxy compounds were investigated in CHO cells expressing individual subtypes of muscarinic receptors and compared with 4-butyloxy analogues.
KEY RESULTS:
The 4-hexyloxy derivatives were found to bind muscarinic receptors with micromolar affinity and antagonized the functional response to carbachol with a potency ranging from 30 nM at M1 to 4 μM at M3 receptors. Under washing conditions to reverse antagonism, the half-life of their antagonistic action ranged from 1.7 h at M2 to 5 h at M5 receptors.
CONCLUSIONS AND IMPLICATIONS:
The 4-hexyloxy derivatives were found to be potent long-acting M1-preferring antagonists. In view of current literature, M1-selective antagonists may have therapeutic potential for striatal cholinergic dystonia, delaying epileptic seizure after organophosphate intoxication or relieving depression. These compounds may also serve as a tool for research into cognitive deficits.
Synthesis of novel and functionally selective non-competitive muscarinic antagonists as chemical probes

Core structure of novel muscarinic antagonists
In collaboration with Barry University novel muscarinic antagonists were developed.
Muscarinic receptors are known to play important biological roles and are drug targets for several human diseases. In a pilot study, novel muscarinic antagonists were synthesized and used as chemical probes to obtain additional information of the muscarinic pharmacophore. The design of these ligands made use of current orthosteric and allosteric models of drug-receptor interactions together with chemical motifs known to achieve muscarinic receptor selectivity. This approach has led to the discovery of several non-competitive muscarinic ligands that strongly bind at a secondary receptor site. These compounds were found to be non-competitive antagonists that completely abolished carbachol activation in functional assays. Several of these compounds antagonized functional response to carbachol with great potency at M1 and M4 than at the rest of receptor subtypes.
Binding of N-methylscopolamine to the extracellular domain of muscarinic acetylcholine receptors

Binding of the orthosteric antagonist N-methylscpolamine to N419 and E175 of the extracellular domain of M2 muscarinic receptor via hydrogen bonds.
This article analyzes in atomistic detail binding of orthosteric ligands to the muscarinic receptors
Interaction of orthosteric ligands with extracellular domain was described at several aminergic G protein-coupled receptors, including muscarinic acetylcholine receptors. The orthosteric antagonists quinuclidinyl benzilate (QNB) and N-methylscopolamine (NMS) bind to the binding pocket of
the muscarinic acetylcholine receptor formed by transmembrane α-helices. We show that high concentrations of either QNB or NMS slow down dissociation of their radiolabeled species from all five subtypes of muscarinic acetylcholine receptors, suggesting allosteric binding. The affinity of NMS at
the allosteric site is in the micromolar range for all receptor subtypes. Using molecular modelling of the M2 receptor we found that E172 and E175 in the second extracellular loop and N419 in the third extracellular loop are involved in allosteric binding of NMS. Mutation of these amino acids to alanine
decreased affinity of NMS for the allosteric binding site confirming results of molecular modelling. The allosteric binding site of NMS overlaps with the binding site of some allosteric, ectopic and bitopic ligands. Understanding of interactions of NMS at the allosteric binding site is essential for correct
analysis of binding and action of these ligands.
Book: Muscarinic Receptor: From Structure to Animal Models
Dr. Jan Jakubik Institute of Physiology Academy of Sciences and professor. Jaromir Mysliveček Institute of Physiology 1st Faculty of Medicine were approached to write the current list of methods that are used in research of muscarinic receptor as part of a Neuromethods series published by Springer.
Dr. Jan Jakubik Institute of Physiology Academy of Sciences and professor Jaromir Mysliveček Institute of Physiology 1st Faculty of Medicine were approached to write the current list of methods that are used in research of muscarinic receptor as part of a Neuromethods series, published by Springer-Verlags. This book provides methods for characterization of muscarinic receptor in crystallography studies that advanced our understanding of structural properties and activation mechanism of muscarinic receptors and are cornerstone in molecular modeling and computer‑based approaches to study muscarinic receptors. Muscarinic Receptor: From Structure to Animal Models guides readers through binding techniques, protocols to investigate molecular properties of muscarinic receptors, protocols to study muscarinic receptors in the central nervous system using autoradiography and PET studies and protocols on animals with knock-out and knock-in muscarinic genes to study role of muscarinic receptors in physiology and behavior. Written in the popular Neuromethods series style, chapters include the kind of detail and key advice from the specialists needed to get successful results in your own laboratory. Concise and easy-to-use, Muscarinic Receptor: From Structure to Animal Models aims to ensure successful results in the further study of this vital field.
Muscarinic receptor: from structure to animal models (eds. Myslivecek, J. & Jakubik, J.)

Towards predictive docking at aminergic G-protein coupled receptors.
Procedure described in this paper represents a possible way to predict interactions of antagonists with aminergic GPCRs.
G protein-coupled receptors (GPCRs) are hard to crystallize. However, attempts to predict their structure have boomed as a result of advancements in crystallographic techniques. This trend has allowed computer-aided molecular modeling of GPCRs. We analyzed the performance of four molecular modeling programs in pose evaluation of re-docked antagonists / inverse agonists to 11 original crystal structures of aminergic GPCRs using an induced fit-docking procedure. AutoDock and Glide were used for docking. AutoDock binding energy function, GlideXP, Prime MM-GB/SA, and YASARA binding function were used for pose scoring. Root mean square deviation (RMSD) of the best pose ranged from 0.09 to 1.58 Å, and median RMSD of the top 60 poses ranged from 1.47 to 3.83 Å. However, RMSD of the top pose ranged from 0.13 to 7.33 Å and ranking of the best pose ranged from the 1st to 60th out of 60 poses. Moreover, analysis of ligand-receptor interactions of top poses revealed substantial differences from interactions found in crystallographic structures. Bad ranking of top poses and discrepancies between top docked poses and crystal structures render current simple docking methods unsuitable for predictive modeling of receptor-ligand interactions. Prime MM-GB/SA optimized for 3NY9 by multiple linear regression did not work well at 3NY8 and 3NYA, structures of the same receptor with different ligands. However, 9 of 11 trajectories of molecular dynamics simulations by Desmond of top poses converged with trajectories of crystal structures. Key interactions were properly detected for all structures. This procedure also worked well for cross-docking of tested β2-adrenergic antagonists. Thus, this procedure represents a possible way to predict interactions of antagonists with aminergic GPCRs.
Comparison of studied receptors

Cross-sections through molecular surface of β‑adrenergic (green), D3 dopamine (yellow), H1 histamine (red) and muscarinic (blue) receptors and their binding sites with bound antagonists. Orientation: extracellular side is up, TM VI and TM VII are in front.
Lipid-based Diets Improve Muscarinic Neurotransmission in the Hippocampus of Transgenic APPswe/PS1dE9 Mice
Even short-term feeding of transgenic mice with chow containing specific lipid-based dietary supplements can influence markers of cholinergic synapses and rectify impaired muscarinic signal transduction that develops in transgenic mice.
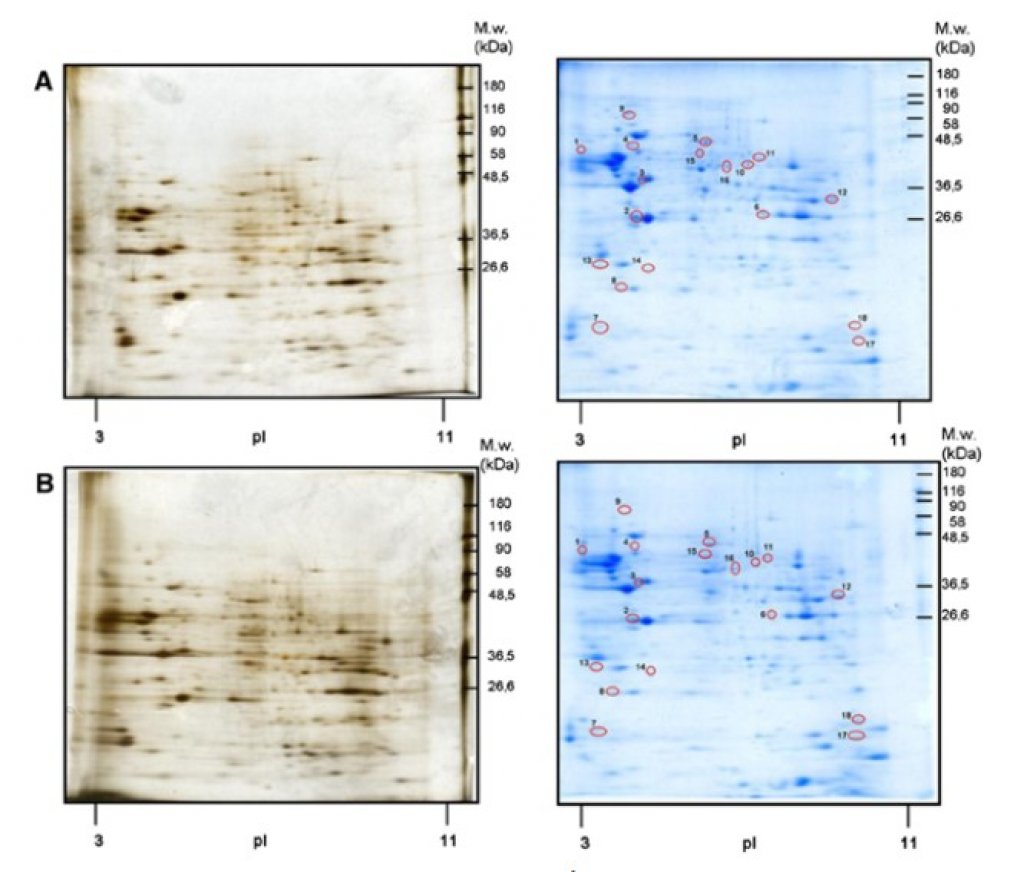
Transgenic APPswe/PS1dE9 mice modelling Alzheimer’s disease demonstrate ongoing accumulation of β-amyloid fragments resulting in formation of amyloid plaques that starts at the age of 4-5 months. Buildup of β-amyloid fragments is accompanied by impairment of muscarinic transmission that becomes detectable at this age, well before the appearance of cognitive deficits that manifest around the age of 12 months. We have recently demonstrated that long-term feeding of trangenic mice with specific isocaloric fish oil-based diets improves specific behavioral parameters. Now we report on the influence of short-term feeding (3 weeks) of three isocaloric diets supplemented with Fortasyn (containing fish oil and ingredients supporting membrane renewal), the plant sterol stigmasterol together with fish oil, and stigmasterol alone on markers of cholinergic neurotransmission in the hippocampus of 5-month-old transgenic mice and their wild-type littermates. Transgenic mice fed normal diet demostrated increase in ChAT activity and attenuation of carbachol-stimulated GTP-γ35S binding compared to wild-type mice. None of the tested diets compared to control diet influenced the activities of ChAT, AChE, BuChE, muscarinic receptor density or carbachol-stimulated GTP-γ35S binding in wild-type mice. In contrast, all experimental diets increased the potency of carbachol in stimulating GTP-γ35S binding in trangenic mice to the level found in wild-type animals. Only the Fortasyn diet increased markers of cholinergic synapses in transgenic mice. Our data demonstrate that even short-term feeding of transgenic mice with chow containing specific lipid-based dietary supplements can influence markers of cholinergic synapses and rectify impaired muscarinic signal transduction that develops in transgenic mice.
Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms
Both classical and atypical agonists activate hM1 receptors by the same molecular switch that involves D71 in the second transmembrane helix. The principal difference among the studied agonists is rather in the way they interact with D105 in the orthosteric binding site.
Randáková, A., Dolejší, E., Rudajev, V., Zimčík, P., Doležal, V., El-Fakahany, E.E. et al. Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms. Pharmacol Res 97, 27-39 (2015).
Graphical abstract

Abstract
We mutated key amino acids of the human variant of the M1 muscarinic receptor that target ligand binding, receptor activation, and receptor-G protein interaction. We compared the effects of these mutations on the action of two atypical M1 functionally preferring agonists (N-desmethylclozapine and xanomeline) and two classical non-selective orthosteric agonists (carbachol and oxotremorine). Mutations of D105 in the orthosteric binding site and mutation of D99 located out of the orthosteric binding site decreased affinity of all tested agonists that was translated as a decrease in potency in accumulation of inositol phosphates and intracellular calcium mobilization. Mutation of D105 decreased the potency of the atypical agonist xanomeline more than that of the classical agonists carbachol and oxotremorine. Mutation of the residues involved in receptor activation (D71) and coupling to G-proteins (R123) completely abolished the functional responses to both classical and atypical agonists. Our data show that both classical and atypical agonists activate hM1 receptors by the same molecular switch that involves D71 in the second transmembrane helix. The principal difference among the studied agonists is rather in the way they interact with D105 in the orthosteric binding site. Furthermore, our data demonstrate a key role of D105 in xanomeline wash-resistant binding and persistent activation of hM1 by wash-resistant xanomeline.
Long-Term Activation upon Brief Exposure to Xanomleline Is Unique to M1 and M4 Subtypes of Muscarinic Acetylcholine Receptors
Our results show commonalities of xanomeline reversible and wash-resistant binding and short-time activation among the five muscarinic receptor subtypes. However long-term receptor activation takes place in full only at hM1 and hM4 receptors. Moreover xanomeline displays higher efficacy at hM1 and hM4 receptors in primary phasic intracellular calcium release. These findings suggest the existence of particular activation mechanisms specific to these two receptors.
Xanomeline is an agonist endowed with functional preference for M1/M4 muscarinic acetylcholine receptors. It also exhibits both reversible and wash-resistant binding to and activation of these receptors. So far the mechanisms of xanomeline selectivity remain unknown. To address this question we employed microfluorometric measurements of intracellular calcium levels and radioligand binding to investigate differences in the short- and long-term effects of xanomeline among muscarinic receptors expressed individually in Chinese hamster ovary cells. 1/ One-min exposure of cells to xanomeline markedly increased intracellular calcium at hM1 and hM4, and to a lesser extent at hM2 and hM3 muscarinic receptors for more than 1 hour. 2/ Unlike the classic agonists carbachol, oxotremorine, and pilocarpine 10-min exposure to xanomeline did not cause internalization of any receptor subtype. 3/ Wash-resistant xanomeline selectively prevented further increase in intracellular calcium by carbachol at hM1 and hM4 receptors. 4/ After transient activation xanomeline behaved as a long-te rm antagonist at hM5 receptors. 5/ The antagonist N-methylscopolamine (NMS) reversibly blocked activation of hM1 through hM4 receptors by xanomeline. 6/ NMS prevented formation of xanomeline wash-resistant binding and activation at hM2 and hM4 receptors and slowed them at hM1, hM3 and hM5 receptors. Our results show commonalities of xanomeline reversible and wash-resistant binding and short-time activation among the five muscarinic receptor subtypes. However long-term receptor activation takes place in full only at hM1 and hM4 receptors. Moreover xanomeline displays higher efficacy at hM1 and hM4 receptors in primary phasic intracellular calcium release. These findings suggest the existence of particular activation mechanisms specific to these two receptors.
Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors

Simulation of molecular dynamic of methoctramine association with M2 receptors. Three stages of molecular dynamics are displayed: Initial (left), transient (middle) and final (right). Extracellular part of the M2 receptor is up and TM IV and V are in fron
Methoctramine high-affinity binding to the M2 receptors involves simultaneous interaction with both the orthosteric and the allosteric binding sites. Lysine 523 in the third extracellular loop of the M3 receptors forms a hydrogen bond with glutamate 219 of the second extracellular loop that hinders methoctramine binding to the allosteric site at this receptor subtype.
Jakubík, J., Zimčík, P., Randáková, A., Fuksová, K., El-Fakahany, E.E. & Doležal, V. Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors. Mol Pharmacol 86, 180-92 (2014).
Methoctramine (N,N’-bis[6-[[(2-methoxyphenyl)-methyl]hexyl]-1,8-octane] diamine) is an M2-selective competitive antagonist of muscarinic acetylcholine receptors and exhibits allosteric properties at high concentrations. To reveal the molecular mechanisms of methoctramine binding and selectivity we took advantage of reciprocal mutations of the M2 and M3 receptors in the second and third extracellular loops that are involved in the binding of allosteric ligands. To this end we performed measurements of kinetics of the radiolabeled antagonists N-methylscopolamine (NMS) in the presence of methoctramine and its precursors, fluorescence energy transfer between green fluorescent protein-fused receptors and an Alexa-555-conjugated precursor of methoctramine, and simulation of molecular dynamics of methoctramine association with the receptor. We confirm the hypothesis that methoctramine high-affinity binding to the M2 receptors involves simultaneous interaction with both the orthosteric binding site and the allosteric binding site located between the second and third extracellular loops. Methoctramine can bind solely with low affinity to the allosteric binding site on the extracellular domain of NMS-occupied M2 receptors by interacting primarily with glutamate 175 in the second extracellular loop. In this mode, methoctramine physically prevents dissociation of NMS from the orthosteric binding site. Our results also demonstrate that lysine 523 in the third extracellular loop of the M3 receptors forms a hydrogen bond with glutamate 219 of the second extracellular loop that hinders methoctramine binding to the allosteric site at this receptor subtype. Impaired interaction with the allosteric binding site manifests as low-affinity binding of methoctramine at the M3 receptor.
Simulation of molecular dynamic of methoctramine association with M2 receptors.

Three stages of molecular dynamics are displayed: Initial (left), transient (middle) and final (right). Extracellular part of the M2 receptor is up and TM IV and V are in front. Backbone of the receptor is colored by position in red to white to blue gradient. Side chains of D103, E172, E175, N404 and Y430 are displayed. Cyan – carbon, blue – nitrogen, green – carbon of methoctramine, red – oxygen; yellow – hydrogen bonds.
Our new paper: Uncoupling of M1 muscarinic receptor/G protein interaction by amyloid β1-42
Janíčková H, Rudajev V, Zimčík P, Jakubík J, Tanila H, El-Fakahany EE, and Doležal V (2013) Neuropharmacology 67: 272-283.
Abstract
The overproduction of β‑amyloid (Aβ) fragments in transgenic APPswe/PS1dE9 mice results in formation of amyloid deposits in the cerebral cortex and hippocampus starting around four months of age and leading to cognitive impairment much later. We have previously found an age and transgene‑ dependent weakening of muscarinic receptor-mediated transmission that was not present in young (6e10-week-old) animals but preceded both amyloid deposits and cognitive deficits. Now we investigated immediate and prolonged in vitro effects of non-aggregated Aβ1‑42 on coupling of individual muscarinic receptor subtypes expressed in CHO (Chinese hamster ovary) cells and their underlying mechanisms. Immediate application of 1 μM Aβ1‑42 had no effect on the binding of the muscarinic antagonist N‑methylscopolamine or the agonist carbachol. In contrast, 4-day treatment of CHO cells expressing the M1 muscarinic receptor with 100 nM Aβ1‑42 significantly changed the binding characteristics of the muscarinic agonist carbachol and reduced the extent of the M1 receptor‑stimulated breakdown of phosphatidylinositol while it did not demonstrate overt toxic effects. The treatment had no influence on the expression of either G-proteins or muscarinic receptors. In concert, we found no change in the gene expression of muscarinic receptor subtypes and gene or protein expression of the Gs, Gq/11, and Gi/o G-proteins in the cerebral cortex of young adult APPswe/PS1dE9 mice that demonstrate high concentrations of soluble Aβ1‑42 and impaired muscarinic receptor-mediated G-protein activation. Our results provide strong evidence that the initial injurious effects of Aβ1‑42 on M1 muscarinic receptor-mediated transmissionis is due to compromised coupling of the receptor with Gq/11 G‑protein.
Our new paper: On homology modeling of the M2 muscarinic acetylcholine receptor subtype

Model of M2 receptor from side- (left) and extracellular- (right) view shows 7 blue α helices spanning the membrane, bound antagonists quinuclidinyl bezilate (green) and amino acids contributing to major intramolecular interactions.
Jakubík J, Randáková A, and Doležal V (2013) J Comput Aided Mol Des 27: 525-538.
Abstract
Twelve homology models of the human M2 muscarinic receptor using different sets of templates have been designed using the Prime program or the modeller program and compared to crystallographic structure (PDB:3UON). The best models were obtained using single template of the closest published structure, the M3 muscarinic receptor (PDB:4DAJ). Adding more (structurally distant) templates led to worse models. Data document a key role of the template in homology modeling. The models differ substantially. The quality checks built into the programs do not correlate with the RMSDs to the crystallographic structure and cannot be used to select the best model. Re-docking of the antagonists present in crystallographic structure and relative binding energy estimation by calculating MM/GBSA in Prime and the binding energy function in YASARA suggested it could be possible to evaluate the quality of the orthosteric binding site based on the prediction of relative binding energies. Although estimation of relative binding energies distinguishes between relatively good and bad models it does not indicate the best one. On the other hand, visual inspection of the models for known features and knowledge-based analysis of the intramolecular interactions allows an experimenter to select overall best models manually.