Čím se zabýváme
Studujeme muskarinové receptory pro acetylcholin (mAChRs), které patří do skupiny receptorů spřažených s G-proteiny (GPCRs) a snažíme se porozumět jejich složitému mechanismu signalizace a způsobu, jakým inter-agují s různými ligandy. Naším hlavním záměrem je prozkoumat potenciál těchto receptorů v léčbě neurologických a psychiatrických onemocnění a také onemocnění dýchacích cest.

Výzkumné projekty
• Muskarinové receptory pro acetylcholin (mAChRs)
Zabýváme se jednotlivými podtypy muskarinivých receptorů (M1–M5), zkoumáme jejich roli v centrálním a periferním nervovém systému a jejich signalizaci v onemocněních jako například Alzheimerova choroba, schizofrenie, nebo astma (Szczurowska et al. 2023, Dolejsi et al. 2024).
• Alosterická modulace receptorů
Zkoumáme vazbu látek do takzvaných alosterických oblastí receptoru, které se liší od klasického (ortosterického) vazebného místa, vedoucí k modulaci aktivity receptorů (Jakubik et al. 2020, Jakubik et El-Fakahany 2020, Jakubik et al. 2019).
• Cílená signalizace
Různí agonisté stabilizují v závislosti na své struktuře různé konformace receptoru (Randakova et al. 2020), a tím aktivují jen některé signální dráhy v buňce (tento jev je znám jako “funkční selektivita” nebo “signalizační bias”) (Randakova et Jakubik 2021).
• Cholesterol, Neurosteroidy, a Neuroactivní steroidy
Studujeme jak membránový cholesterol, neurosteroidy a steroidní hormony alostericky modulují muskarinové a další GPCR. (Szczurowska et al. 2022, Jakubik et El-Fakahany 2021).
• Vývoj léčiv a léčebných postupů
Snažíme se identifikovat a vyvíjet látky s potenciálem pro léčbu Alzheimerovy choroby, schizofrenie, Parkinsonovy nemoci, epilepsie, deprese, závislostí, astmatu, chronické obstrukční plicní nemoci a bolesti. (Janous-kova-Randakova et al. 2025, Randakova et al. 2020, Randakova et al. 2018).
Významné výsledky
Naše studie přinesly řadu klíčových zjištění, jež posouvají hranice současných vědeckých znalostí.
• Teoretické vymezení signalizačního biasu na úrovni jednotlivých podtypů G-proteinů
Podrobně jsme prokázali, že agonisté stabilizují různé konformace receptorů, vedoucí k odlišným biologickým účinkům. Tento koncept přesahuje zjednodušený pohled na aktivaci receptorů spřažených s G–proteiny (GPCR). Vyvinuli a charakterizovali jsme nové muskarinové agonisty, které vykazují signalizační bias – tedy preferenční aktivaci specifických drah zprostředkovaných jednotlivými G–proteiny (například agonisty recep-torů M2 a M4, které selektivně aktivují Gi proteiny a preferenčně tak inhibují tvorbu cAMP) (Nelic et al. 2024, Randakova et al. 2020).
Cílená signalizace představuje slibnou strategii pro vývoj léčiv, která dosahují požadovaných terapeutických účinků a zároveň minimalizují nežádoucí vedlejší účinky tím, že se vyhýbají aktivaci nežádoucích signálních drah. Naše práce také přináší fúzní proteiny (receptor spojený s konkrétní podjednotkou Gα) jako účinný nástroj pro přesnou analýzu biasu agonistů, zejména u nekanonických drah. Tento přístup zajišťuje přesný poměr receptoru ku α-podjednotce G-proteinu 1:1 a omezuje přístup kompetujících Gα podjednotek(Randakova et al. 2021).

• Rozvoj teorie agonismu u receptor-efektorových systémů
Rozvinuli jsme operační model agonismu (OMA), který slouží jako standard pro hodnocení síly účinku agonistů nezávisle na systému. Tento model má však má své limity a úskalí (Jakubik et al. 2023). K jejich překonání jsme vyvinuli robustní dvoustupňovou analytickou metodu pro aplikaci operačního modelu na experimentální data (Jakubik et al. 2019). Dále jsme odvodili rovnice pro operační model alosterické modulace agonismu (Jakubik et al. 2020). Odvodili jsme explicitní rovnice pro operační model agonismu dimerů receptorů (OMARD), který je použitelný pro jakýkoliv receptor se dvěma ortosterickými vazebnými místy (Jakubik et Randakova, 2022). I přes vyšší komplexnost při prokládání experimentálních dat umožňují tato vylepšení dosud používaného modelu získat mnohem přesnější výsledky.
Dále jsme se zabývaly úlohou guanosindifosfátu (GDP) při aktivaci receptorů. Našli jsme důkazy, že negativní kooperativita mezi vazbou agonisty a GDP představuje alternativní způsob měření účinnosti agonistů, který dokáže odhalit mechanistické rozdíly nepozorovatelné ve studiích vazby GTP (Jakubik et al. 2011).

• Antagonisté muskarinových receptorů s dlouhodobým účinkem
Náš výzkum se podrobně zabýval unikátní neodmyvatelnou vazbou xanomelinu na mAChR, přičemž bylo pro-kázáno, že tato vazba může vést k dlohodobé aktivaci nebo inhibici receptoru v závislosti na jeho podtypu (Jakubik et al. 2002) a že tento jev je ovlivněn O-alkylovým postranním řetězcem ligandu interagujícím s membránou (Jakubik et al. 2004). Rovněž jsme syntetizovali nové dlouhodobě působící antagonisty přednostně působící na M1 receptor, obsahující hexyloxy řetězec, který je zodpovědný za jeho pomalou disociaci z receptoru (Randakova et al. 2018). Detailní porozumění mechanismu dlouhodobé interakce ligand–receptor je zásadní pro návrh léčiv s prodlouženou dobou účinku, což může vést ke snížení denní dávky a zlepšit adherenci pacientů k terapeutickému režimu.

• Popis zásadní úlohy vestibulu vazebné kapsy ve funkci receptoru
Náš model „dvou tandemově uspořádaných vazebných míst“ pro klasické antagonisty nabízí nová vysvětlení celé řady jevů, které byly dříve připisovány izomerizaci receptoru, včetně mechanismu účinku dlouhodobě působícího muskarinového antagonisty tiotropia. (Jakubik et al. 2017, Jakubik et al. 2014, Jakubik et al. 2000).

• Objevení společného nekanonického vazebného místa pro cholesterol
Popsali jsme nekanonické vazebné místo pro cholesterol, které se nachází na transmembránové helixu 6 (TM6) a to jak u muskarinových, tak u opioidních receptorů (Chetverikov et al. 2025, preprint). Tento objev navazuje na zjištění, že neurosteroidy a steroidní hormony alostericky modulují mAChR již při fyziologicky významných nanomolárních koncentracích (Dolejsi et al. 2021a) Toto vazebné místo je odlišné od běžného alosterického místa nacházejícího se v extracelulárních smyčkách a je orientováno směrem k membráně (Dolejsi et al. 2021b). Rozdílná citlivost různých GPCR k modulaci aktivity cholesterolem může představovat molekulární základ pro funkční selektivitu. Tato zjištění naznačují, že některé z dobře známých příznivých účinků léčby steroidy u poruch CNS mohou být částečně způsobeny jejich alosterickou modulací mAChR, kromě jejich účinků na jiné receptory, jako jsou NMDA a GABA receptory (Szczurowska et al. 2023).

• Objev alosterických agonistů
Identifikovali a charakterizovali jsme řadu alosterických modulátorů, které mohou působit sami jako agonisté. Naše raná práce ukázala, že alosterické modulátory, jako je alkuronium, gallamin a strychnin, mohou vyvolat funkční odpovědi zprostředkované G-proteinem i v nepřítomnosti ortosterických agonistů, představující nový mechanismus aktivace receptoru (Jakubik et al. 1996). Také jsme zjistili, že alosterické modulátory mohou pozitivně ovlivnit afinitu muskarinových receptorů k agonistům, včetně acetylcholinu, což bylo v té době průlomové zjištění (Jakubik et al. 1997). Tento objev otevřel nové možnosti pro zesílení cholinergního přenosu fyziologickým způsobem, tedy pouze v místě a čase, kdy je receptor aktivován endogenním acetylcholinem (Jakubik et El-Fakahany, 2020).

Translační význam
Naše výzkumy mají významný potenciál pro využití ve vývoji nových léčiv a v terapii onemocnění: Bezpečnější léčiva: Biased ligandy (např. M2 funkčně selektivní agonisté) mohou sloužit jako neopioidní analgetika s nižším rizikem nežádoucích účinků.
Cílená terapie:
Alzheimerova choroba: Pozitivní alosterická modulace receptoru M1 neurosteroidy.
Schizofrenie: Alosterická modulace receptorů M1/M4 s nižším rizikem kognitivních nežádoucích účinků.
Parkinsonova choroba: Inhibice M1/M4 pohlavními hormony a jinými steroidy.
Epilepsie: Zapojení M2/M4 do záchvatů navozených steroidy.
Závislosti: Inhibice M5 k omezení vyhledávání drog.
Dlouhodobě působící léčiva: Ligandy rezistentní na odmytí / dlouhodobě účinné ligandy mohou snížit frekvenci dávkování a zlepšit adherenci pacientů.
Tkáňově specifická léčiva: Tkáňově specifická distribuce jednotlivých podtypů G-proteinů a jejih selektivní modulace umožnuje lokalizované účinky léčiva.
Chemické sondy: Naše nové sloučeniny pomáhají mapovat funkce receptorů a podporují vývoj nových léčiv.
Používané metody
Náš multidisciplinární přístup zahrnuje:
Vazebné studie
Vazba radioligandů k určení afinity a kinetiky testovaných látek
FRET (Förster resonance energy transfer) ke sledování interakcí ligand–receptor v reálném čase.
Polarizace anizotropie fluorescence – podobně jako FRET, ale bez nutnosti modifikace receptoru.
Funkční studie
Vazba GTPγS ke studiu aktivace G-proteinů.
Měření uvolňování intracelulárního vápníku a akumulace inositolfosfátů pro analýzu PLC signální dráhy.
Meření inhibice cAMP (např. GloSensor™).
Uvolňování acetylcholinu v řezech mozku k studiu presynaptických receptorů.
BRET (Bioluminiscence resonance energy transfer) testy pro sledování aktivace G-proteinů v reálném čase v živých buňkách.
Molekulární biologie
Cílená mutageneze k identifikaci klíčových aminokyselinových zbytků.
Konstrukce fúzních proteinů pro analýzu specifických signálních drah.
Výpočetní metody
Molekulární modelování a simulace dynamiky k analýze vazby a struktury receptoru.
Analýza dat
Analýza dat pomocí specializovaného analytického softwaru, pokročilého modelování a prokládání křivek v jazyce Python a statistického zpracování v prostředí R.
Vhledy do operačního modelu agonismu dimerickáých receptorů

Vlevo, homodimer sestává ze dvou identických receptorů R vážících jednu molekulu agonisty A, každý s rovnovážnou disociační konstantou KA. Vpravo se heterodimer skládá ze dvou různých receptorů R, které vážou jednu molekulu agonisty A, každý s rovnovážnou
Jakubík, J.; Randáková, A. Insights into the Operational Model of Agonism of Receptor Dimers. Expert Opin. Drug Discov. 2022, 17, 1181–1191, doi:10.1080/17460441.2023.2147502. Zvaný přehled.
- Určení přesného pořadí účinnosti a potence agonistů je nepostradatelné při objevování nových selektivních agonistů.
- Operační model agonismu (OMA) je současným standardem.
Mnoho receptorů funguje jako oligomery, které vyžadují rozšíření klasického modelu OMA. - Rozšíření modelu OMA o faktory sklonu křivek funkční odezvy poskytuje jednoduché rovnice, které lze snadno přizpůsobit experimentálním datům, ale výsledky mohou být zavádějící.
- Rozšíření modelu OMA o kooperativní faktory poskytuje přesné, ale složité rovnice funkční odezvy.
Rozšíření molelu OMA o kooperativní faktory by mělo být preferováno před rozšířením faktorem sklonu.
Úvod: Přesné seřazení účinnosti a potence agonistů je zásadní pro objevování nových selektivních agonistů. Pro účely klasifikace agonistů nezávislé na systému se standardem stal operační model agonismu (OMA). Mnoho receptorů funguje jako oligomery, což činí funkční odpovědi složitějšími, což vyžaduje rozšíření původní OMA.
Obsah: Byly odvozeny explicitní rovnice operačního modelu agonismu receptorových dimerů (OMARD). OMARD lze aplikovat na jakýkoli receptor, který má dvě ortosterická místa. Chování OMARD bylo analyzováno, aby se prokázala jeho složitost a vztah k experimentálním datům. Vlastnosti rovnic OMARD a OMA byly porovnány, aby se demonstrovaly jejich klady a zápory.
Expertní názor: Rozšíření OMA o faktory sklonu poskytuje jednoduché rovnice funkční odezvy, které lze snadno přizpůsobit experimentálním datům, ale výsledky mohou být nepřesné. Také takové rovnice nemohou pojmout křivky ve tvaru zvonu. Explicitní rovnice OMARD poskytují přesné výsledky, ale jsou složité a zdlouhavé, aby se vešly do experimentálních dat. Všechny provozní modely používají vzájemně závislé parametry, které jsou překážkou v armatuře.
Funkčně selektivní a směřovaní agonisté muskarinových receptorů

Směřovaná signalizace muskarinových receptorů
Randáková, A.; Jakubík, J. Functionally Selective and Biased Agonists of Muscarinic Receptors. Pharmacol. Res. 2021, 169, 105641, doi:10.1016/j.phrs.2021.105641.
Zvaný přehled.
Narušení cholinergní signalizace prostřednictvím muskarinových receptorů je spojeno s různými patologiemi, jako je Alzheimerova choroba nebo schizofrenie. Selektivní muskarinoví agonisté mají terapeutický potenciál při léčbě diabetu, bolesti nebo Sjögrenova syndromu. Ortosterické vazebné místo všech podtypů muskarinového receptoru je strukturálně identické, takže vývoj selektivních agonistů na bázi afinity je prakticky nemožný. Někteří agonisté jsou však funkčně selektivní; aktivují pouze podskupinu receptorů nebo signálních drah. Jiné mohou stabilizovat specifické konformace receptoru, což vede k nejednotné modulaci jednotlivých signálních drah (směřovaní agonisté). Funkčně selektivní a směřovaní agonisté představují slibný přístup v selektivní aktivaci jednotlivých subtypů muskarinových receptorů. V této práci rekapitulujeme chemické struktury, vazbu na receptor a specifické konformace pro agonisty v dosud známých funkčně selektivních a směrovaných agonistů muskarinových receptorů v kontextu jejich složité intracelulární signalizace. Dále se zabýváme perspektivou možného použití směřovaných agonistů pro tkáňově a orgánově specifickou aktivaci muskarinových receptorů.
Nikotinové acetylcholinové receptory exprimované ve striatálních interneuronech inhibují striatální aktivitu a kontrolují chování závislé na striatu.

Behaviorální testy provedené u jednotlivých kohort myší. Je uvedeno pořadí testů v každé kohortě a přibližný věk testovaných zvířat. Každý měsíc věku označujeme šipkou; každá šipka může obsahovat 4 tečky představující 4 týdny v měsíci. Na této časové ose
Abbondanza, A.; Ribeiro Bas, I.; Modrak, M.; Capek, M.; Minich, J.; Tyshkevich, A.; Naser, S.; Rangotis, R.; Houdek, P.; Sumova, A.; et al. Nicotinic Acetylcholine Receptors Expressed by Striatal Interneurons Inhibit Striatal Activity and Control Striatal-Dependent Behaviors. J. Neurosci. 2022, 42, 2786–2803, doi:10.1523/JNEUROSCI.1627-21.2022.
Velké množství nikotinových receptorů pro acfetylcholin (nAChR) je exprimováno ve striatu, oblasti mozku, která je klíčová při kontrole chování. Složitost vztahů receptorů s různými funkcemi brání našemu pochopení mechanismů, kterými striatální acetylcholin moduluje chování. Zaměřili jsme se na roli reletivně malé populace nAChR obsahujících beta2 podjednotku. Identifikovali jsme neuronální typy exprimující tyto receptory a určili jsme jejich vliv na kontrolu explorativního chování, chování podobného úzkosti, učení a citlivosti na stimulanty. Další experimenty ukázaly, že tyto změny byly spojeny s celkově zvýšenou aktivitou striatálních neuronů. Malá populace nikotinových receptorů tedy představuje zajímavý cíl pro modulaci odezvy na stimulační léky a další chování založené na striatálním stavu.
Acetylcholin je důležitým modulátorem striatální aktivity a je životně důležitý pro kontrolu striatálně závislého chování, včetně motorických a kognitivních funkcí. Navzdory tomuto významu nejsou mechanismy určující, jak acetylcholin ovlivňuje striatální signalizaci, stále plně pochopeny. Zejména je málo známo o úloze nikotinových receptorů (nAChR) exprimovaných striatálními interneurony. V této studii jsme použili metodu FISH k určení, které typy neuronů exprimují nejrozšířenější beta2 nikotinovou podjednotku v myším striatu. Naše data podporují běžný názor, že exprese nAChR je většinou omezena na striatální interneurony. Překvapivě však byly cholinergní interneurony identifikovány jako populace s nejvyšší expresí beta2 nikotinové podjednotky. Abychom prozkoumali funkční význam nAChR obsahujících beta2 v striatálních interneuronech, odstranili jsme je injekcí vektoru AAV-Cre do striata samců myší beta2-flox/flox. Delece vedla ke změnám v několika behaviorálních doménách, jmenovitě ke zvýšenému chování podobnému úzkosti, snížení poměru sociability, deficitu v diskriminačním učení a zvýšené amfetaminem indukované hyperlokomoci a expresi c-Fos u myší s delecí beta2. Další kolokalizační analýza ukázala, že zvýšená exprese c-Fos byla přítomna jak ve středních výběžkovitých neuronech, tak u předpokládaných striatálních interneuronů. Tato studie dochází k závěru, že přestože jsou relativně vzácné, nAChR obsahující beta2 jsou primárně exprimovány v striatálních neuronech cholinergními interneurony a hrají významnou roli v chování.
Neurosteroidy a steroidní hormony jsou alosterické modulátory muskarinových receptorů

Dolejší, E.; Szánti-Pintér, E.; Chetverikov, N.; Nelic, D.; Randáková, A.; Doležal, V.; Kudová, E.; Jakubík, J. Neurosteroids and Steroid Hormones Are Allosteric Modulators of Muscarinic Receptors. Neuropharmacology 2021, 199, 108798, doi:10.1016/j.neuropharm.2021.108798.
- Některé neurosteroidy a steroidní hormony se váží na muskarinové acetylcholinové receptory s afinitou 100 nM nebo vyšší
- Steroidy působící v nanomolárních koncentracích představují nový farmakofor alosterických modulátorů muskarinových receptorů
- Kortikosteron a progesteron alostericky modulují muskarinové receptory ve fyziologicky relevantních koncentracích
Bylo zjištěno, že membránový cholesterol váže a moduluje funkci mnoha receptorů spojených s G-proteinem, včetně muskarinových acetylcholinových receptorů. Zkoumali jsme vazbu 20 steroidních sloučenin včetně neurosteroidů a steroidních hormonů na muskarinové receptory. Kortikosteron, progesteron a některé neurosteroidy se váží na muskarinové receptory s afinitou 100 nM nebo vyšší. Ukázali jsme, že kortikosteron a progesteron alostericky modulují funkční odpověď muskarinových receptorů na acetylcholin ve fyziologicky relevantních koncentracích. Může hrát roli při kontrole stresu nebo v těhotenství, kdy hladiny těchto hormonů dramaticky oscilují. Allosterická modulace muskarinových receptorů přes místo vázající cholesterol představuje nový farmakologický přístup k onemocněním spojeným se změněnou cholinergní signalizací. Dále jsme stanovili vztah mezi strukturou a aktivitou pro alosterické modulátory muskarinových receptorů na bázi steroidů.
The operational model of allosteric modulation of pharmacological agonism

The cubic ternary complex model of allosteric modulation of receptor activation. Inactive, R, and active, R*, states of the receptor, K<sub>ACT</sub> receptor activation constant, K<sub>A,/sub> and K<sub>B</sub> affinities.
Proper determination of agonist efficacy is indispensable in the evaluation of agonist selectivity and bias to activation of specific signalling pathways. The operational model of pharmacological agonism is a useful means for achieving this goal.
Monod et al. (1963) originally introduced the concept of allosterism. Since then the concept of allosterism extended to many various fields of research spanning from DNA expression via metabolism to ion channels and G-protein coupled receptors. Allosteric ligands bind to a site that is distinct from the orthosteric site on a receptor. An orthosteric and allosteric ligand can bind to the receptor concurrently and form a ternary complex where they reciprocally modulate the binding affinity of each other. Moreover, the binding of an allosteric modulator may also affect the efficacy of an orthosteric agonist in eliciting a functional response.
Allosteric modulators are an intensively studied group of receptor ligands because of their selectivity and preservation of physiological space-time pattern of the signals they modulate. We analysed the operational model of allosterically-modulated agonism (OMAM) including modulation by allosteric agonists. Several parameters of OMAM are inter-dependent. We derived equations describing mutual relationships among parameters of the functional response and OMAM. We present a workflow for the robust fitting of OMAM to experimental data using derived equations.
Novel M2-Selective, Gi-Biased Agonists of Muscarinic Acetylcholine Receptors

Obecné schéma spřahování GPCR s jednotlivými podtypy G-proteinů nebo arrestiny po aktivaci agonistou tradičního typu (vlevo) a agonistou preferujícím aktivaci Gi-proteinu (vpravo). AC, adenylát cykláza; PLC fosfolipáza C.
Vyvinuli jsme nové agonisty muskarinových receptorů, jako frmakofor pro vývoj nových analgetik nezpůsobujících návyk jako opiáty, nebo oslabení imunity jako steroidní analgetika. Tato muskarinová analgetika by nezpůsobovala vedlejší účinky jako je inkontinence, nadměrné slinění a pocení a další.
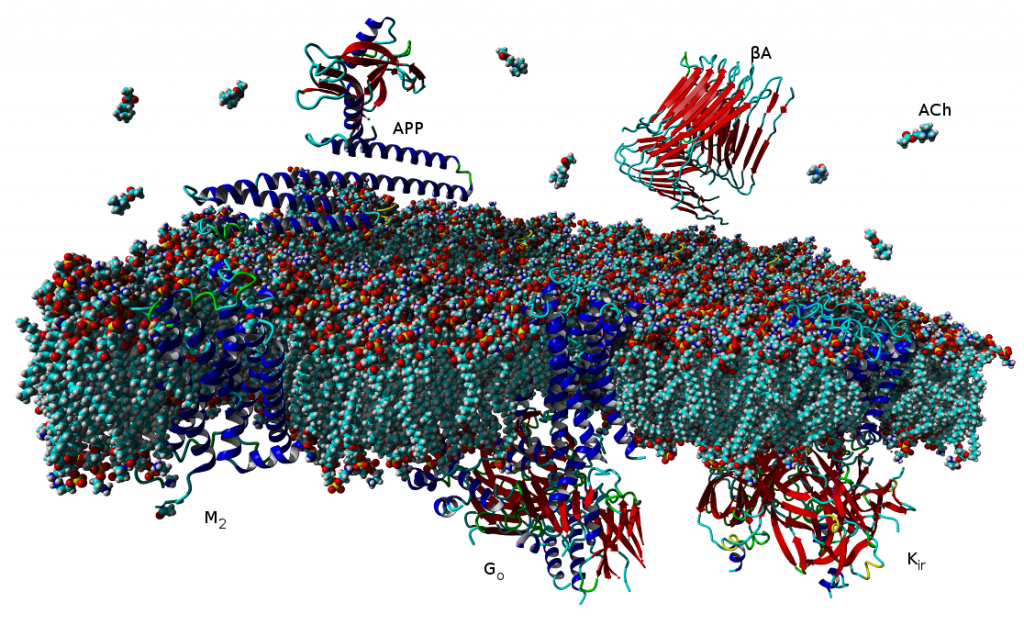
V současné době více než třetina všech léčiv na trhu cílí na receptory v membránách buněk, které jsou spřaženy s vnitrobuněčnými signálními G‑proteiny (tzv. GPCR). Tento typ membránových receptorů se podílí na řízení široké škály fyziologických procesů, počínaje zpracováním smyslových podnětů, přes regulaci chování a nálady, hormonální a imunitní odpovědi až po řízení autonomních funkcí a buněčné proliferace. Proto tyto receptory obecně představují jeden z nejdůležitějších farmakoterapeutických cílů. Tradiční agonisté GPCR aktivují v buňkách obvykle vícero signálních drah, což může mít za následek řadu vedlejších účinků. Látky vedoucí výlučně k aktivaci jedné signální dráhy představují novou generaci vysoce specifických léčiv s méně vedlejšími účinky. Velké úsilí bylo dosud věnováno výzkumu látek vedoucích buď k aktivaci G‑proteinových drah, nebo k signalizační kaskádě přes β‑arrestiny.
V naší laboratoři se zaměřujeme na studium muskarinových acetylcholinových receptorů, které patří k typickým představitelům GPCR. V naší publikaci představujeme zcela nový typ agonistů těchto receptorů, kteří dokážou specificky modulovat signální dráhy na úrovni spřahování receptoru s jednotlivými podtypy G‑proteinů (Obrázek 1A). Zmíněné látky působí výhradně na signální dráhu přes protein Gi, která vede k inhibici adenylát cyklázy a tím ke snížení hladiny cAMP v buňce (Obrázek 1B). Díky tomu jsou tyto látky funkčně selektivní pro muskarinové receptory M2 a M4, jež představují jeden z farmakologických cílů při léčbě bolesti. Nově objevení agonisté mohou vést k vývoji nových analgetik nezpůsobujících návyk jako opiáty, nebo oslabení imunity jako steroidní analgetika. Tato muskarinová analgetika by nezpůsobovala vedlejší účinky zprostředkované aktivací signální dráhy přes protein Gq, jako je inkontinence, nadměrné slinění a pocení a další.
Agonist-specific conformations of the M2 muscarinic acetylcholine receptor assessed by molecular dynamics

Structurally diverse agonists for a given receptor induce receptor conformations specific to each structural family. These agonist-specific conformations can lead to non-uniform modulation of signalling pathways. This preferential orientation of signalling of a given receptor towards a subset of its signal transducers is termed signalling bias. This property may be employed to develop drugs that selectively produce desired effects while avoiding side effects associated with activation of unwanted signalling pathways. We modelled conformations of the M2 receptor specific to individual agonists, including the newly developed Gi-biased agonists.
Binding of muscarinic ligands, both antagonists and agonists, and their effects on the conformation of the M2 acetylcholine receptor were modelled in silico and compared to experimental data. After docking of antagonists to the M2 receptor in an inactive conformation (3UON, 5ZK3, 5ZKB or 5ZKB) and agonists in an active conformation (4MQS) 100 ns of conventional molecular dynamics (MD) followed by 500 ns of accelerated MD was run. Conventional MD revealed ligand-specific interactions with the receptor. Antagonists stabilized the receptor in an inactive conformation during accelerated MD. The receptor in complex with various agonists attained different conformations specific to individual agonists. The magnitude of TM6 movement correlated with agonist efficacy at non-preferential Gs pathway. The shape of the intracellular opening where the receptor interacts with a G-protein was different for the classical agonist carbachol, super-agonist iperoxo and Gi/o-biased partial agonists JR-6 and JR-7, being compatible with experimentally observed agonist bias at the G-protein level. Moreover, wash-resistant binding of the unique agonist xanomeline associated with interaction with membrane lipids was formed during accelerated MD. Thus, accelerated MD is suitable for modelling of ligand-specific receptor binding and receptor conformations that is essential for the design of experiments aimed at the identification of the secondary binding sites and understanding molecular mechanisms underlying receptor activation.
Applications and limitations of fitting of the operational model to determine relative efficacies of agonists

Graphical Abstract
Proper determination of agonist efficacy is essential in the assessment of agonist selectivity and signalling bias. Agonist efficacy is a relative term that is dependent on the system in which it is measured, especially being dependent on receptor expression level. In this work, we analyse limits and pitfalls of fitting OM to experimental data.
Proper determination of agonist efficacy is essential in the assessment of agonist selectivity and signalling bias. Agonist efficacy is a relative term that is dependent on the system in which it is measured, especially being dependent on receptor expression level. The operational model (OM) of functional receptor agonism is a useful means for the determination of agonist functional efficacy using the maximal response to agonist and ratio of agonist functional potency to its equilibrium dissociation constant (KA) at the active state of the receptor. However, the functional efficacy parameter τ is inter-dependent on two other parameters of OM; agonist’s KA and the highest response that could be evoked in the system by any stimulus (EMAX). Thus, fitting of OM to functional response data is a tricky process. In this work, we analyse pitfalls of fitting OM to experimental data and propose a rigorous fitting procedure where KA and EMAX are derived from the half-efficient concentration of agonist and apparent maximal responses obtained from a series of functional response curves. Subsequently, OM with fixed KA and EMAX is fitted to functional response data to obtain τ. The procedure was verified at M2 and M4 muscarinic receptors fused with the G15 G-protein α-subunit. The procedure, however, is applicable to any receptor-effector system.
Role of membrane cholesterol in differential sensitivity of muscarinic receptor subtypes to persistently bound xanomeline

Our new publication in journal Neuropharmacology in which we demonstrate that membrane cholesterol plays an important and subtype-specific role in activation of muscarinic acetylcholine receptors. To our knowledge, this is the first demonstration of pharmacological selectivity due to differences in receptor-membrane interactions at any GPCR. The possibility to achieve pharmacological selectivity based on receptor-membrane interactions changes our view on the molecular basis of pharmacological selectivity and opens new ways for the development of a novel pharmaceutics.
Xanomeline (3-(Hexyloxy)-4-(1-methyl-1,2,5,6-tetrahydropyridin-3-yl)-1,2,5-thiadiazole) is a muscarinic agonist that is considered to be functionally selective for the M1/M4 receptor subtypes. Part of xanomeline binding is resistant to washing. Wash-resistant xanomeline activates muscarinic receptors persistently, except for the M5 subtype. Mutation of leucine 6.46 to isoleucine at M1 or M4 receptors abolished persistent activation by wash-resistant xanomeline. Reciprocal mutation of isoleucine 6.46 to leucine at the M5 receptor made it sensitive to activation by wash-resistant xanomeline. Lowering of membrane cholesterol made M1 and M4 mutants and M5 wild type receptors sensitive to activation by wash-resistant xanomeline. Molecular docking revealed a cholesterol binding site in the groove between transmembrane helices 6 and 7. Molecular dynamics showed that interaction of cholesterol with this binding site attenuates receptor activation. We hypothesize that differences in cholesterol binding to this site between muscarinic receptor subtypes may constitute the basis for xanomeline apparent functional selectivity and may have notable therapeutic implications. Differences in receptor-membrane interactions, rather than in agonist-receptor interactions, represent a novel possibility to achieve pharmacological selectivity. Our findings may be applicable to other G protein coupled receptors.

Cholesterol binding to the intracellular half of TM6 of wt (left) and L376I mutant (right) M1 receptor based on crystal structure 5CXV (Thal et al., 2016) is shown. Orientation, extracellular side up, TM6 front. Colours: magenta, surface of TM5, TM6 and TM7, yellow, surface of R365, L376 and I376; cyan, carbon; white, hydrogen; red, oxygen.
Novel long-acting antagonists of muscarinic ACh receptors

Graphical Abstract
In this study, we have developed new potent and long-acting antagonists of muscarinic acetylcholine receptors with a potential therapeutic use.
BACKGROUND AND PURPOSE:
The aim of this study was to develop potent and long-acting antagonists of muscarinic ACh receptors. The 4-hexyloxy and 4-butyloxy derivatives of 1-[2-(4-oxidobenzoyloxy)ethyl]-1,2,3,6-tetrahydropyridin-1-ium were synthesized and tested for biological activity. Antagonists with long-residence time at receptors are therapeutic targets for the treatment of several neurological and psychiatric human diseases. Their long-acting effects allow for reduced daily doses and adverse effects.
EXPERIMENTAL APPROACH:
The binding and antagonism of functional responses to the agonist carbachol mediated by 4-hexyloxy compounds were investigated in CHO cells expressing individual subtypes of muscarinic receptors and compared with 4-butyloxy analogues.
KEY RESULTS:
The 4-hexyloxy derivatives were found to bind muscarinic receptors with micromolar affinity and antagonized the functional response to carbachol with a potency ranging from 30 nM at M1 to 4 μM at M3 receptors. Under washing conditions to reverse antagonism, the half-life of their antagonistic action ranged from 1.7 h at M2 to 5 h at M5 receptors.
CONCLUSIONS AND IMPLICATIONS:
The 4-hexyloxy derivatives were found to be potent long-acting M1-preferring antagonists. In view of current literature, M1-selective antagonists may have therapeutic potential for striatal cholinergic dystonia, delaying epileptic seizure after organophosphate intoxication or relieving depression. These compounds may also serve as a tool for research into cognitive deficits.
Synthesis of novel and functionally selective non-competitive muscarinic antagonists as chemical probes

Core structure of novel muscarinic antagonists
Ve spolupráci s Barry University byli vyvinuti noví antagonisté muskarinových receptorů.
Muscarinic receptors are known to play important biological roles and are drug targets for several human diseases. In a pilot study, novel muscarinic antagonists were synthesized and used as chemical probes to obtain additional information of the muscarinic pharmacophore. The design of these ligands made use of current orthosteric and allosteric models of drug-receptor interactions together with chemical motifs known to achieve muscarinic receptor selectivity. This approach has led to the discovery of several non-competitive muscarinic ligands that strongly bind at a secondary receptor site. These compounds were found to be non-competitive antagonists that completely abolished carbachol activation in functional assays. Several of these compounds antagonized functional response to carbachol with great potency at M1 and M4 than at the rest of receptor subtypes.
Binding of N-methylscopolamine to the extracellular domain of muscarinic acetylcholine receptors

Binding of the orthosteric antagonist N-methylscpolamine to N419 and E175 of the extracellular domain of M2 muscarinic receptor via hydrogen bonds.
This article analyzes in atomistic detail binding of orthosteric ligands to the muscarinic receptors
Interaction of orthosteric ligands with extracellular domain was described at several aminergic G protein-coupled receptors, including muscarinic acetylcholine receptors. The orthosteric antagonists quinuclidinyl benzilate (QNB) and N-methylscopolamine (NMS) bind to the binding pocket of
the muscarinic acetylcholine receptor formed by transmembrane α-helices. We show that high concentrations of either QNB or NMS slow down dissociation of their radiolabeled species from all five subtypes of muscarinic acetylcholine receptors, suggesting allosteric binding. The affinity of NMS at
the allosteric site is in the micromolar range for all receptor subtypes. Using molecular modelling of the M2 receptor we found that E172 and E175 in the second extracellular loop and N419 in the third extracellular loop are involved in allosteric binding of NMS. Mutation of these amino acids to alanine
decreased affinity of NMS for the allosteric binding site confirming results of molecular modelling. The allosteric binding site of NMS overlaps with the binding site of some allosteric, ectopic and bitopic ligands. Understanding of interactions of NMS at the allosteric binding site is essential for correct
analysis of binding and action of these ligands.
Kniha: Muscarinic Receptor: From Structure to Animal Models
Dr. Jan Jakubík z Fyziologického ústavu Akademie věd a prof. Jaromír Mysliveček z Fyziologického ústavu 1. LF UK byly osloveni k sepsání aktuálního přehledu metod, které se používají při výzkumu muskarinových receptorů, jako součásti série Neuromethods, která je jednou z významných edic nakladatelství Springer.
Dr. Jan Jakubík z Fyziologického ústavu Akademie věd a prof. Jaromír Mysliveček z Fyziologického ústavu 1. LF UK byly osloveni k sepsání aktuálního přehledu metod, které se používají při výzkumu muskarinových receptorů, jako součásti série Neuromethods, která je jednou z významných edic nakladatelství Springer. Podařilo se sestavit knihu, která zahrnuje základní popis muskarinových receptorů pomocí krystalografických studií, vazebných studií, autoradiografických stanovení a PET zobrazení. Kniha se zabývá i studiem alosterického ovivňování muskarinových receptorů a na příkladech modelů myší s vyřazenými geny muskarinových receptorů (metoda knock-out) ukazuje jejich využití ve studiu fyziologických pochodů a chování. Dá se předpokládat, že by mohla být pomůckou pro všechny vědce, kteří potřebují ke svému výzkumu studovat vlastnosti muskarinových receptorů.
Muscarinic receptor: from structure to animal models (eds. Myslivecek, J. & Jakubik, J.)

Towards predictive docking at aminergic G-protein coupled receptors.
Procedure described in this paper represents a possible way to predict interactions of antagonists with aminergic GPCRs.
G protein-coupled receptors (GPCRs) are hard to crystallize. However, attempts to predict their structure have boomed as a result of advancements in crystallographic techniques. This trend has allowed computer-aided molecular modeling of GPCRs. We analyzed the performance of four molecular modeling programs in pose evaluation of re-docked antagonists / inverse agonists to 11 original crystal structures of aminergic GPCRs using an induced fit-docking procedure. AutoDock and Glide were used for docking. AutoDock binding energy function, GlideXP, Prime MM-GB/SA, and YASARA binding function were used for pose scoring. Root mean square deviation (RMSD) of the best pose ranged from 0.09 to 1.58 Å, and median RMSD of the top 60 poses ranged from 1.47 to 3.83 Å. However, RMSD of the top pose ranged from 0.13 to 7.33 Å and ranking of the best pose ranged from the 1st to 60th out of 60 poses. Moreover, analysis of ligand-receptor interactions of top poses revealed substantial differences from interactions found in crystallographic structures. Bad ranking of top poses and discrepancies between top docked poses and crystal structures render current simple docking methods unsuitable for predictive modeling of receptor-ligand interactions. Prime MM-GB/SA optimized for 3NY9 by multiple linear regression did not work well at 3NY8 and 3NYA, structures of the same receptor with different ligands. However, 9 of 11 trajectories of molecular dynamics simulations by Desmond of top poses converged with trajectories of crystal structures. Key interactions were properly detected for all structures. This procedure also worked well for cross-docking of tested β2-adrenergic antagonists. Thus, this procedure represents a possible way to predict interactions of antagonists with aminergic GPCRs.
Comparison of studied receptors

Cross-sections through molecular surface of β‑adrenergic (green), D3 dopamine (yellow), H1 histamine (red) and muscarinic (blue) receptors and their binding sites with bound antagonists. Orientation: extracellular side is up, TM VI and TM VII are in front.
Lipid-based Diets Improve Muscarinic Neurotransmission in the Hippocampus of Transgenic APPswe/PS1dE9 Mice
Even short-term feeding of transgenic mice with chow containing specific lipid-based dietary supplements can influence markers of cholinergic synapses and rectify impaired muscarinic signal transduction that develops in transgenic mice.
Transgenic APPswe/PS1dE9 mice modelling Alzheimer’s disease demonstrate ongoing accumulation of β-amyloid fragments resulting in formation of amyloid plaques that starts at the age of 4-5 months. Buildup of β-amyloid fragments is accompanied by impairment of muscarinic transmission that becomes detectable at this age, well before the appearance of cognitive deficits that manifest around the age of 12 months. We have recently demonstrated that long-term feeding of trangenic mice with specific isocaloric fish oil-based diets improves specific behavioral parameters. Now we report on the influence of short-term feeding (3 weeks) of three isocaloric diets supplemented with Fortasyn (containing fish oil and ingredients supporting membrane renewal), the plant sterol stigmasterol together with fish oil, and stigmasterol alone on markers of cholinergic neurotransmission in the hippocampus of 5-month-old transgenic mice and their wild-type littermates. Transgenic mice fed normal diet demostrated increase in ChAT activity and attenuation of carbachol-stimulated GTP-γ35S binding compared to wild-type mice. None of the tested diets compared to control diet influenced the activities of ChAT, AChE, BuChE, muscarinic receptor density or carbachol-stimulated GTP-γ35S binding in wild-type mice. In contrast, all experimental diets increased the potency of carbachol in stimulating GTP-γ35S binding in trangenic mice to the level found in wild-type animals. Only the Fortasyn diet increased markers of cholinergic synapses in transgenic mice. Our data demonstrate that even short-term feeding of transgenic mice with chow containing specific lipid-based dietary supplements can influence markers of cholinergic synapses and rectify impaired muscarinic signal transduction that develops in transgenic mice.
Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms
Both classical and atypical agonists activate hM1 receptors by the same molecular switch that involves D71 in the second transmembrane helix. The principal difference among the studied agonists is rather in the way they interact with D105 in the orthosteric binding site.
Randáková, A., Dolejší, E., Rudajev, V., Zimčík, P., Doležal, V., El-Fakahany, E.E. et al. Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms. Pharmacol Res 97, 27-39 (2015).
Graphical abstract

Abstract
We mutated key amino acids of the human variant of the M1 muscarinic receptor that target ligand binding, receptor activation, and receptor-G protein interaction. We compared the effects of these mutations on the action of two atypical M1 functionally preferring agonists (N-desmethylclozapine and xanomeline) and two classical non-selective orthosteric agonists (carbachol and oxotremorine). Mutations of D105 in the orthosteric binding site and mutation of D99 located out of the orthosteric binding site decreased affinity of all tested agonists that was translated as a decrease in potency in accumulation of inositol phosphates and intracellular calcium mobilization. Mutation of D105 decreased the potency of the atypical agonist xanomeline more than that of the classical agonists carbachol and oxotremorine. Mutation of the residues involved in receptor activation (D71) and coupling to G-proteins (R123) completely abolished the functional responses to both classical and atypical agonists. Our data show that both classical and atypical agonists activate hM1 receptors by the same molecular switch that involves D71 in the second transmembrane helix. The principal difference among the studied agonists is rather in the way they interact with D105 in the orthosteric binding site. Furthermore, our data demonstrate a key role of D105 in xanomeline wash-resistant binding and persistent activation of hM1 by wash-resistant xanomeline.
Long-Term Activation upon Brief Exposure to Xanomleline Is Unique to M1 and M4 Subtypes of Muscarinic Acetylcholine Receptors
Our results show commonalities of xanomeline reversible and wash-resistant binding and short-time activation among the five muscarinic receptor subtypes. However long-term receptor activation takes place in full only at hM1 and hM4 receptors. Moreover xanomeline displays higher efficacy at hM1 and hM4 receptors in primary phasic intracellular calcium release. These findings suggest the existence of particular activation mechanisms specific to these two receptors.
Xanomeline is an agonist endowed with functional preference for M1/M4 muscarinic acetylcholine receptors. It also exhibits both reversible and wash-resistant binding to and activation of these receptors. So far the mechanisms of xanomeline selectivity remain unknown. To address this question we employed microfluorometric measurements of intracellular calcium levels and radioligand binding to investigate differences in the short- and long-term effects of xanomeline among muscarinic receptors expressed individually in Chinese hamster ovary cells. 1/ One-min exposure of cells to xanomeline markedly increased intracellular calcium at hM1 and hM4, and to a lesser extent at hM2 and hM3 muscarinic receptors for more than 1 hour. 2/ Unlike the classic agonists carbachol, oxotremorine, and pilocarpine 10-min exposure to xanomeline did not cause internalization of any receptor subtype. 3/ Wash-resistant xanomeline selectively prevented further increase in intracellular calcium by carbachol at hM1 and hM4 receptors. 4/ After transient activation xanomeline behaved as a long-te rm antagonist at hM5 receptors. 5/ The antagonist N-methylscopolamine (NMS) reversibly blocked activation of hM1 through hM4 receptors by xanomeline. 6/ NMS prevented formation of xanomeline wash-resistant binding and activation at hM2 and hM4 receptors and slowed them at hM1, hM3 and hM5 receptors. Our results show commonalities of xanomeline reversible and wash-resistant binding and short-time activation among the five muscarinic receptor subtypes. However long-term receptor activation takes place in full only at hM1 and hM4 receptors. Moreover xanomeline displays higher efficacy at hM1 and hM4 receptors in primary phasic intracellular calcium release. These findings suggest the existence of particular activation mechanisms specific to these two receptors.
Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors

Simulation of molecular dynamic of methoctramine association with M2 receptors. Three stages of molecular dynamics are displayed: Initial (left), transient (middle) and final (right). Extracellular part of the M2 receptor is up and TM IV and V are in fron
Methoctramine high-affinity binding to the M2 receptors involves simultaneous interaction with both the orthosteric and the allosteric binding sites. Lysine 523 in the third extracellular loop of the M3 receptors forms a hydrogen bond with glutamate 219 of the second extracellular loop that hinders methoctramine binding to the allosteric site at this receptor subtype.
Jakubík, J., Zimčík, P., Randáková, A., Fuksová, K., El-Fakahany, E.E. & Doležal, V. Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors. Mol Pharmacol 86, 180-92 (2014).
Methoctramine (N,N‘-bis[6-[[(2-methoxyphenyl)-methyl]hexyl]-1,8-octane] diamine) is an M2-selective competitive antagonist of muscarinic acetylcholine receptors and exhibits allosteric properties at high concentrations. To reveal the molecular mechanisms of methoctramine binding and selectivity we took advantage of reciprocal mutations of the M2 and M3 receptors in the second and third extracellular loops that are involved in the binding of allosteric ligands. To this end we performed measurements of kinetics of the radiolabeled antagonists N-methylscopolamine (NMS) in the presence of methoctramine and its precursors, fluorescence energy transfer between green fluorescent protein-fused receptors and an Alexa-555-conjugated precursor of methoctramine, and simulation of molecular dynamics of methoctramine association with the receptor. We confirm the hypothesis that methoctramine high-affinity binding to the M2 receptors involves simultaneous interaction with both the orthosteric binding site and the allosteric binding site located between the second and third extracellular loops. Methoctramine can bind solely with low affinity to the allosteric binding site on the extracellular domain of NMS-occupied M2 receptors by interacting primarily with glutamate 175 in the second extracellular loop. In this mode, methoctramine physically prevents dissociation of NMS from the orthosteric binding site. Our results also demonstrate that lysine 523 in the third extracellular loop of the M3 receptors forms a hydrogen bond with glutamate 219 of the second extracellular loop that hinders methoctramine binding to the allosteric site at this receptor subtype. Impaired interaction with the allosteric binding site manifests as low-affinity binding of methoctramine at the M3 receptor.
Simulation of molecular dynamic of methoctramine association with M2 receptors.

Three stages of molecular dynamics are displayed: Initial (left), transient (middle) and final (right). Extracellular part of the M2 receptor is up and TM IV and V are in front. Backbone of the receptor is colored by position in red to white to blue gradient. Side chains of D103, E172, E175, N404 and Y430 are displayed. Cyan – carbon, blue – nitrogen, green – carbon of methoctramine, red – oxygen; yellow – hydrogen bonds.
Naše nová publikace: Rozpřažení interakce mezi M1 muskarinovými receptory a G-proteiny β1-42 amyloidem
Janíčková H, Rudajev V, Zimčík P, Jakubík J, Tanila H, El-Fakahany EE, and Doležal V (2013) Neuropharmacology 67: 272-283.
Abstract
The overproduction of β‑amyloid (Aβ) fragments in transgenic APPswe/PS1dE9 mice results in formation of amyloid deposits in the cerebral cortex and hippocampus starting around four months of age and leading to cognitive impairment much later. We have previously found an age and transgene‑ dependent weakening of muscarinic receptor-mediated transmission that was not present in young (6e10-week-old) animals but preceded both amyloid deposits and cognitive deficits. Now we investigated immediate and prolonged in vitro effects of non-aggregated Aβ1‑42 on coupling of individual muscarinic receptor subtypes expressed in CHO (Chinese hamster ovary) cells and their underlying mechanisms. Immediate application of 1 μM Aβ1‑42 had no effect on the binding of the muscarinic antagonist N‑methylscopolamine or the agonist carbachol. In contrast, 4-day treatment of CHO cells expressing the M1 muscarinic receptor with 100 nM Aβ1‑42 significantly changed the binding characteristics of the muscarinic agonist carbachol and reduced the extent of the M1 receptor‑stimulated breakdown of phosphatidylinositol while it did not demonstrate overt toxic effects. The treatment had no influence on the expression of either G-proteins or muscarinic receptors. In concert, we found no change in the gene expression of muscarinic receptor subtypes and gene or protein expression of the Gs, Gq/11, and Gi/o G-proteins in the cerebral cortex of young adult APPswe/PS1dE9 mice that demonstrate high concentrations of soluble Aβ1‑42 and impaired muscarinic receptor-mediated G-protein activation. Our results provide strong evidence that the initial injurious effects of Aβ1‑42 on M1 muscarinic receptor-mediated transmissionis is due to compromised coupling of the receptor with Gq/11 G‑protein.
Naše nová publikace: Homologní modelování muskarinového receptoru M2 pro acetylcholin

Model M2 receptoru při pohledu z boku (vlevo) a z mimobuněčného prostoru (vpravo) ukazuje 7 modrých α helixů protínajících buněčnou membránu a navázaného antagonistu chinuklidinylbezilát (zelený).
Jakubík J, Randáková A, and Doležal V (2013) J Comput Aided Mol Des 27: 525-538.
Abstract
Twelve homology models of the human M2 muscarinic receptor using different sets of templates have been designed using the Prime program or the modeller program and compared to crystallographic structure (PDB:3UON). The best models were obtained using single template of the closest published structure, the M3 muscarinic receptor (PDB:4DAJ). Adding more (structurally distant) templates led to worse models. Data document a key role of the template in homology modeling. The models differ substantially. The quality checks built into the programs do not correlate with the RMSDs to the crystallographic structure and cannot be used to select the best model. Re-docking of the antagonists present in crystallographic structure and relative binding energy estimation by calculating MM/GBSA in Prime and the binding energy function in YASARA suggested it could be possible to evaluate the quality of the orthosteric binding site based on the prediction of relative binding energies. Although estimation of relative binding energies distinguishes between relatively good and bad models it does not indicate the best one. On the other hand, visual inspection of the models for known features and knowledge-based analysis of the intramolecular interactions allows an experimenter to select overall best models manually.