Insulin is a hormone that helps keep blood glucose levels within a safe range and enables cells to use glucose as their primary energy source. It is produced by β-cells in the islets of Langerhans in the pancreas, which, within the microorgan of the islet, closely cooperate with other hormone-producing cells, particularly α- and δ-cells. Disruption of communication between these cells is among the processes associated with the development of type 2 diabetes. A new study by scientists from IPHYS, published in the journal Comprehensive Physiology, shows that under conditions of early β-cell dysfunction, α-cells can activate a compensatory program that supports residual insulin secretion. Understanding these compensatory mechanisms is important for developing new strategies to protect β-cells and delay the onset of type 2 diabetes.
Why insulin matters and where it is produced
Pancreatic β-cells release insulin after a meal, helping to lower blood glucose levels. In type 2 diabetes, however, their ability to respond to glucose gradually declines. New work from the Laboratory of Pancreatic Islet Research shows that in this early phase, not only β-cells themselves, but the entire pancreatic islet as an interconnected cellular system, plays a key role.
“To study this, we used a mouse model with β-cell-specific deletion of the enzyme NADPH oxidase 4 (Nox4). These mice show impaired glucose-stimulated insulin secretion and exhibit features of early diabetes, a so-called prediabetic state,” explains the corresponding author of the study, Lydie Plecitá-Hlavatá. Detailed analysis of pancreatic islets in these mice revealed an increased proportion of α-cells and a higher number of cells displaying characteristics of both α- and β-cells.
α-cells as emergency support for β-cells
α-cells have long been viewed primarily as the “counterpart” to β-cells, as they produce glucagon, a hormone that raises blood glucose levels. “However, our study shows that when β-cell function is impaired, the behavior of neighboring α-cells changes. They increase the production of glucagon and the local hormone GLP-1 and, through intercellular communication, likely contribute to maintaining residual insulin secretion,” summarizes Plecitá-Hlavatá.
In the islets of the mouse model with β-cell dysfunction, signaling through glucagon and GLP-1 receptors was altered. Intracellular pathways associated with cAMP and calcium regulation – both crucial for insulin secretion -were also enhanced. The study thus demonstrates that β-cell impairment does not lead to isolated failure, but rather triggers a reorganization of communication within the entire islet.
The findings suggest that, under metabolic stress affecting β-cells, the pancreatic islet flexibly rewires communication between different cell types. Although this adaptation cannot fully compensate for β-cell dysfunction, it may help sustain insulin secretion in the early stages of the disease. The study therefore offers a new perspective on prediabetes: it is not merely the failure of a single cell type, but a shift in the function of the entire islet.
The work highlights the importance of a multidisciplinary approach to diabetes research. Instead of focusing on β-cells in isolation, the islet should be understood as a dynamically adapting cellular network. A detailed understanding of regulatory mechanisms and targeting therapies toward compensatory processes within the islets could help better protect β-cells during prediabetes and delay the development of type 2 diabetes. At the same time, the authors note that this is preclinical research conducted in a mouse model, and it will be necessary to verify whether similar mechanisms occur in humans.
The study was carried out in collaboration between the IPHYS and the University of Maribor and was supported by the National Institute for Research of Metabolic and Cardiovascular Diseases CarDia (EXCELES, LX22NPO5104) and the Czech Science Foundation.
Reference: Benáková Š., Holendová B., Dolenšek J., Křivonosková M., Stožer A., Plecitá-Hlavatá L.: Pancreatic Islet Cell Crosstalk: Insight Into α-/β-Cell Compensatory Mechanisms. Comprehensive Physiology 16:e70158 (2026). DOI: 10.1002/cph4.70158. IF = 5.2.
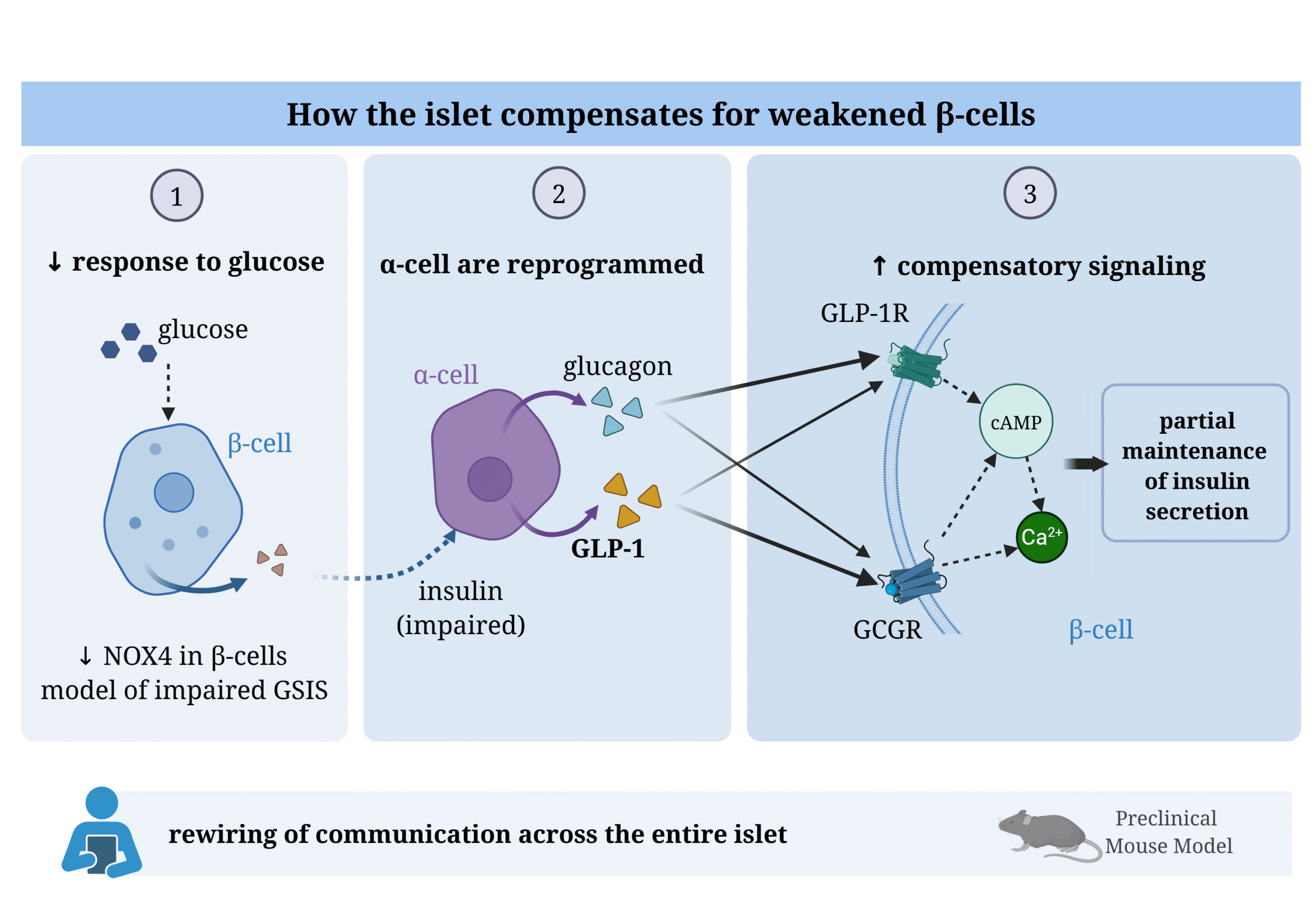
Figure legend: Compensatory remodeling of intercellular communication within the pancreatic islet under impaired β-cell function. (1) β-cell–specific deletion of Nox4 in a mouse model leads to a reduced β-cell response to glucose and impaired glucose-stimulated insulin secretion (GSIS).
(2) The diminished insulin output is associated with remodeling of the α-cell population, including an increased proportion of α-cells, expansion of bihormonal cells, and elevated production of glucagon and GLP-1.
(3) These α-cell–derived paracrine signals act via GLP-1R and GCGR receptors on β-cells, promoting cAMP-dependent and Ca²⁺-dependent signaling pathways, which may partially sustain the secretory capacity of β-cells despite their primary defect. This adaptation reflects a functional reprogramming of communication across the entire islet. However, its relevance to early stages of human diabetes requires further validation.
Abbreviations: NOX4 = NADPH oxidase 4; GSIS = glucose-stimulated insulin secretion; GLP-1 = glucagon-like peptide-1; GLP-1R = GLP-1 receptor; GCGR = glucagon receptor; cAMP = cyklický adenosinmonofosfát; Ca²⁺ = vápenatý ion
{kind=link}